
CRISPR Guide
CRISPR Overview
Bacteria have an interesting adaptive immune response that detects and eliminates foreign DNA, which scientists have modified for a wide range of genome engineering techniques. Collectively, these techniques are referred to as CRISPR (Clustered Regularly Interspaced Short Palindromic Repeat) technologies. Before CRISPR, genome engineering approaches like zinc finger nucleases (ZFNs) or transcription-activator-like effector nucleases (TALENs) required scientists to design and generate a new nuclease pair for every genomic target. Due to its comparative simplicity and adaptability, CRISPR rapidly became the most popular genome engineering approach.
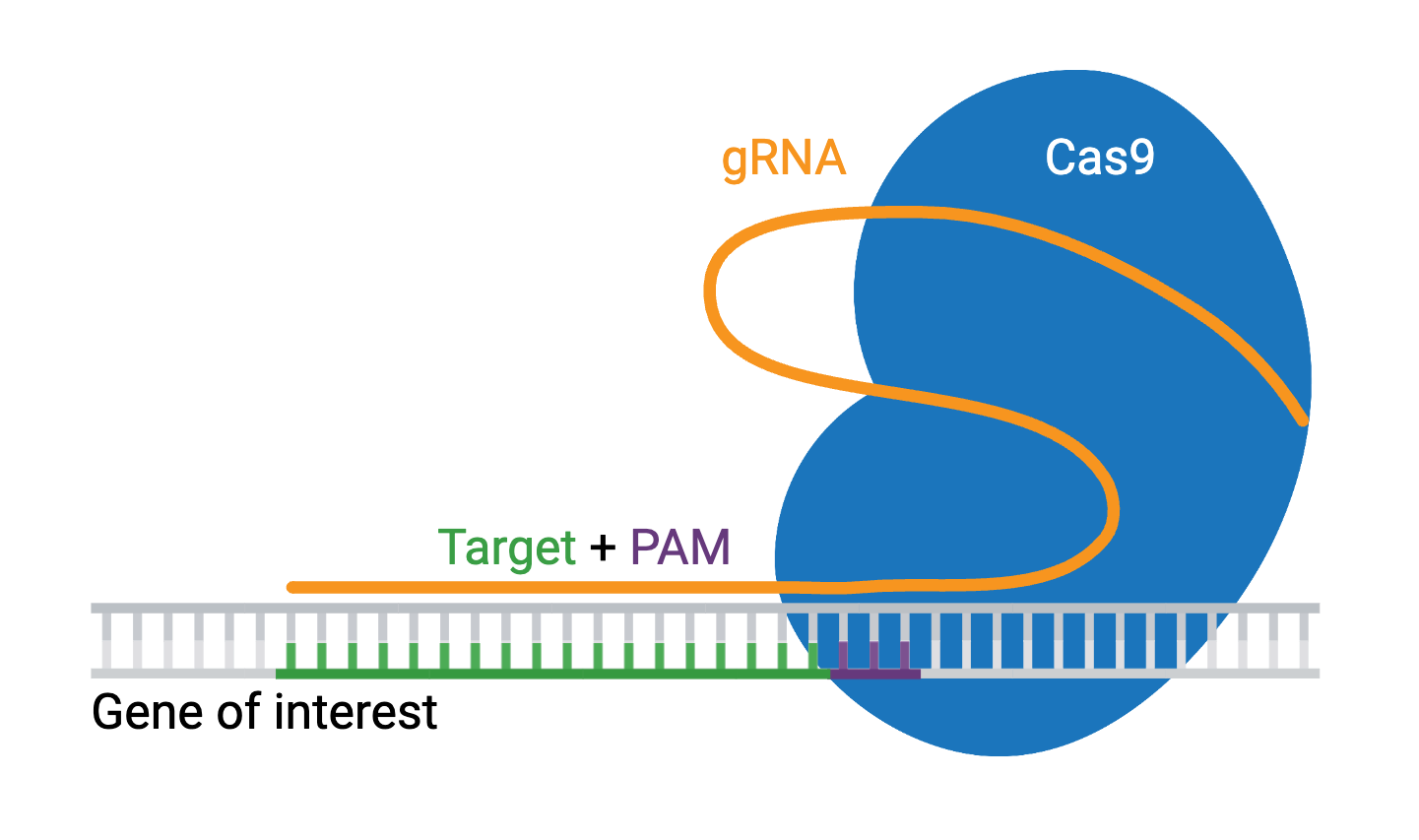
Engineered CRISPR systems contain two components: a guide RNA (gRNA or sgRNA) and a CRISPR-associated endonuclease (Cas enzyme). The gRNA is a short synthetic RNA composed of a scaffold sequence necessary for Cas-binding and a user-defined ∼20-nucleotide spacer that defines the genomic target to be modified. You can therefore change the genomic target of the Cas enzyme by simply changing the target sequence present in the gRNA.
CRISPR was originally employed to knock out target genes in various cell types and organisms, but modifications to various Cas enzymes have extended CRISPR applications to increasingly complex functions, including the ability to selectively activate/repress target genes, purify specific regions of DNA, image DNA in live cells, and precisely edit DNA and RNA. The ease of generating gRNAs makes CRISPR one of the most scalable genome editing technologies, making CRISPR perfect for genome-wide screens.
This guide will provide a basic understanding of CRISPR biology, introduce the various applications of CRISPR, and help you get started using CRISPR in your own research.
CRISPR Basics
In this section, we will introduce the basic mechanisms of CRISPR and some of the ways the CRISPR components are leveraged to achieve specific goals, such as targeting multiple genomic locations or enhancing specificity, efficiency, or flexibility of the Cas enzyme.
Basic Mechanisms of CRISPR
The requirements for a CRISPR knockout assay are simple. You’ll need a Cas enzyme and a gRNA specific to the gene of interest. The genomic target of the gRNA can be any ~20 nucleotide sequence, provided it meets two conditions:
- The sequence is unique compared to the rest of the genome
- The target is present immediately adjacent to a Protospacer Adjacent Motif (PAM)
The PAM sequence (NGG) serves as a binding signal for Cas9, but the exact sequence depends on which Cas protein you use, and there are several to choose from. Many variations of Cas9 exist, along with other Cas enzymes like Cas12a, which are available for specialized applications. Check out our list of additional Cas proteins and PAM sequences.
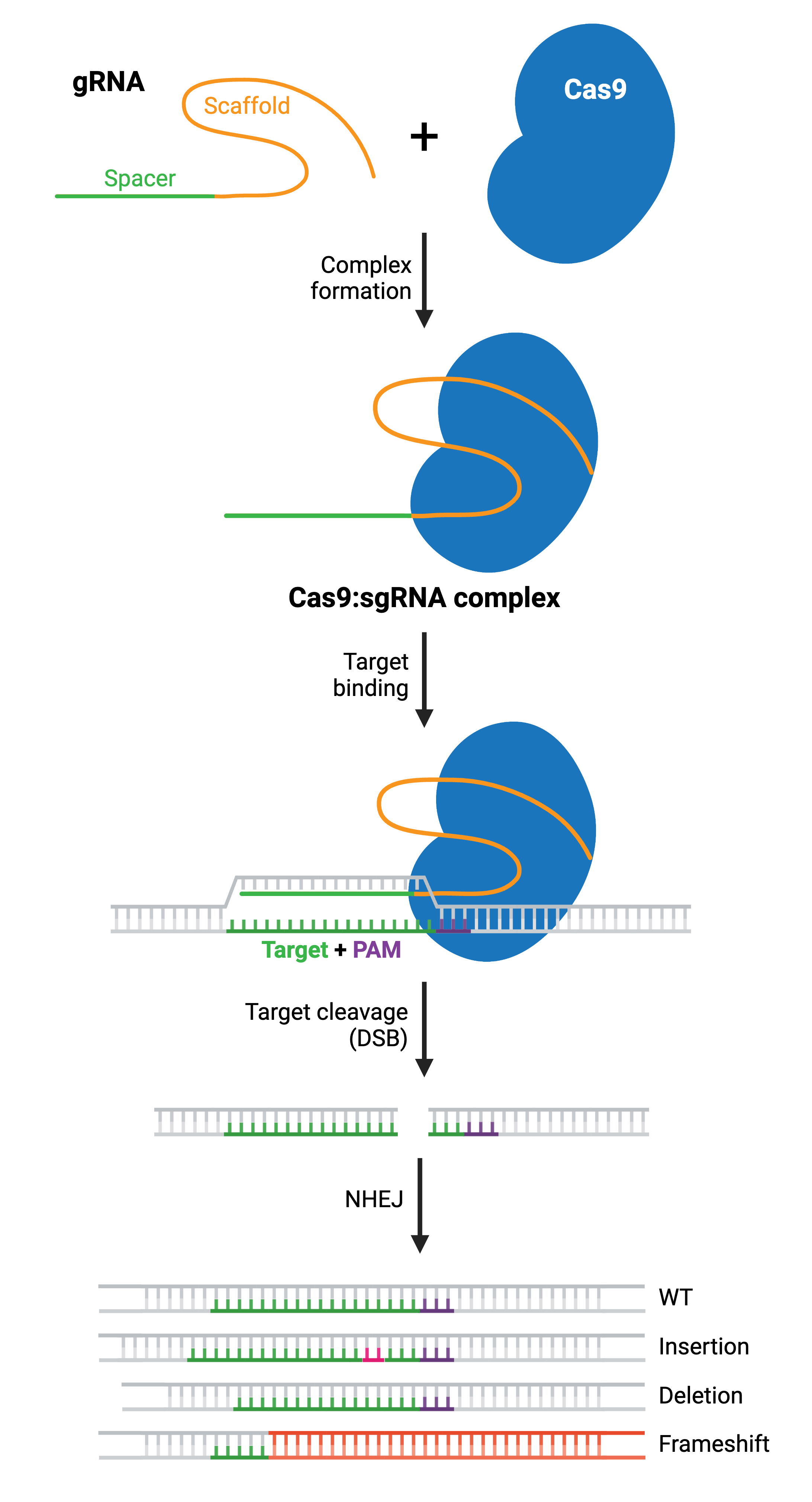
Once you’ve selected your target, you’ll express your gRNA and Cas9 protein, which will form a ribonucleoprotein complex through interactions between the gRNA scaffold and surface-exposed, positively-charged grooves on Cas9. gRNA binding induces a conformational change in Cas9, shifting it into an active, DNA-binding configuration, with the spacer region of the gRNA left free to interact with the target DNA.
Cas9 will only cleave a given locus if the gRNA spacer sequence shares sufficient homology with the target DNA. Once the Cas9-gRNA complex binds a putative DNA target, the seed sequence (8–10 bases at the 3′ end of the gRNA targeting sequence) will begin to anneal to the target DNA. If the seed and target DNA sequences match, the gRNA will continue to anneal to the target DNA in a 3′ to 5′ direction. The location of any potential mismatches matters. Mismatches between the target sequence in the 3′ seed sequence inhibit target cleavage, while mismatches toward the 5′ end distal to the PAM often permit target cleavage.
Generating a Knockout Using CRISPR
Cas9 undergoes a second conformational change upon target binding. In this formation, its nuclease domains, RuvC and HNH, cleave opposite strands of the target DNA. This results in a double-strand break (DSB) within the target DNA, located ∼3–4 nucleotides upstream of the PAM sequence.
The resulting DSB is then repaired by one of two general repair pathways: the efficient but error-prone non-homologous end joining (NHEJ) pathway and the less efficient but high-fidelity homology directed repair (HDR) pathway. HDR relies on copying DNA from a matching template to accurately repair or fill in the missing sequence, and is more commonly used for small precision edits. NHEJ directly ligates the break ends without the need for a homologous template. Both NHEJ and HDR have their uses, but we’ll focus on NHEJ first and come back to HDR.
NHEJ is a naturally occurring repair pathway and is the primary repair mechanism for DSBs. While this pathway is efficient in repairing DSBs, it frequently causes small nucleotide insertions or deletions (indels) at the DSB site. Therefore, a population of cells expressing Cas9 and a gRNA will result in a diverse array of mutations (see: Validate genetic modification). In most cases, NHEJ gives rise to small indels in the target DNA that result in amino acid deletions, insertions, or frameshift mutations leading to premature stop codons within the open reading frame (ORF) of the targeted gene. The ideal result is a loss-of-function mutation within the targeted gene. However, the strength of the knockout phenotype for a given mutant cell must be validated experimentally.
Browse Plasmids: Double-Strand Break (Cut)
Multiplex Genome Engineering
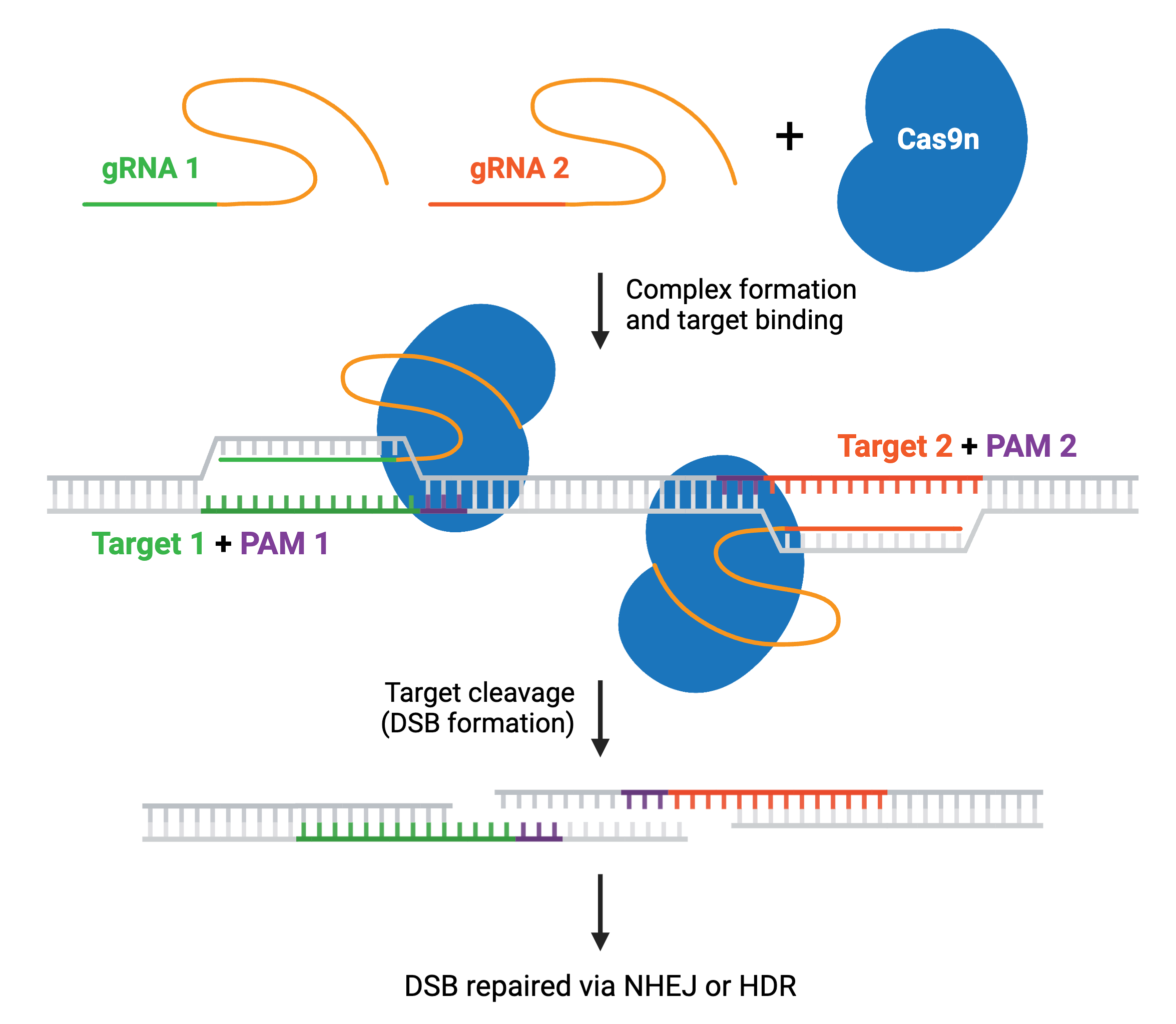
Many CRISPR experiments involve editing more than one gene. In these cases, delivering multiple gRNAs using a single plasmid ensures that all gRNAs will be expressed in the same cell. This is called multiplexing, and increases the likelihood that any cell containing the CRISPR plasmid will have all desired genomic edits carried out by Cas9. Multiplexing applications include editing multiple genes at once; using dual nickases to generate a knockout or gene edit; or using Cas9 to generate large genomic deletions or inversions with two gRNA sites on the same chromosome.
Most multiplex systems enable researchers to target anywhere from 2–7 genetic loci by cloning multiple gRNAs into a single plasmid — though some have achieved targeting in the double digits. These multiplex gRNA vectors can be used to knock out, activate, or repress target genes when paired with the appropriate Cas enzyme. Specific enzymes, such as Cas12a or Cas13, can improve multiplexing efficiency and are discussed in more detail later. Read more about Cas9 multiplexing.
Browse Plasmids: Multiplex gRNA Vectors
Engineering the Cas9 Endonuclease
CRISPR specificity is determined by both the gRNA sequence and the Cas9 enzyme. Ideally, a gRNA targeting sequence will have perfect homology to the target DNA with no homology elsewhere in the genome. Realistically, most gRNA targeting sequences will have additional sites of partial homology throughout the genome, called off-targets, that can impact your experiment. There are many online tools available to help you select an optimized gRNA for your application (see: gRNA Design Software).
In addition to an optimized gRNA design, engineered Cas9s can enhance CRISPR specificity. SpCas9 (from Streptococcus pyogenes) is the most popular Cas endonuclease, with many modified versions engineered to increase specificity and nuclease activity available. Engineered Cas9s have also led to reduced dependency on specific PAM sequences and the creation of new tools.
Nickase and dead Cas9
The two Cas9 nuclease domains, RuvC and HNH, can be altered to reduce cleavage events to a single cut (nick) or no cuts. Cas9 nickase (Cas9n), a D10A mutant of SpCas9, has one active nuclease domain and one inactivated one, allowing it to generate DNA nicks by cutting only one strand. Catalytically inactive “dead” Cas9 (dCas9) contains mutations in both nuclease domains, D10A and H840A, resulting in no cleavage of target DNA.
Cas9n increases target specificity. Since two nickases targeting opposite DNA strands are required to generate a DSB within the target DNA, it’s unlikely that two off-target nicks will be generated close enough to cause a DSB. The nickase system is utilized in many CRISPR systems to achieve specific genetic edits.
The dCas9 molecule binds to target DNA based on the gRNA sequence without cleavage. This means proteins with other functions can be fused to a dCas9 and delivered to specific genomic locations to carry out unique tasks. We’ll discuss more Cas9 fusion tools later in the guide.
Increasing fidelity of Cas9
Researchers have also enhanced specificity by engineering Cas9 into high fidelity Cas9s (hfCas9). There is currently no defined measure of fidelity, so sometimes they will be referred to as increased fidelity enzymes. hfCas9s were developed by mutating specific amino acid residues to reduce off-target editing. Some mutations disrupt interactions between Cas9 and DNA, while others work to increase Cas9’s proofreading capabilities.
No matter the method, increased fidelity enzymes lead to less off-target editing than wild type Cas9. Examples of increased fidelity enzymes, and their mechanisms, include:
- eSpCas9(1.1) — weaken interactions between the HNH/RuvC groove and the non-target DNA strand
- SpCas9-HF1 — disrupt Cas9’s interactions with DNA phosphate backbone
- HypaCas9 — increase Cas9 proofreading and discrimination
- evoCas9 — decrease off-target effects
- xCas9 3.7 — mutations in multiple domains to expand PAM recognition, increase specificity, and decrease off-target effects
- Sniper-Cas9 — less off-target activity; compatible with truncated gRNAs to increase specificity
- SuperFi-Cas9 — increases fidelity with reduced nuclease activity
Cas9 enzymes will likely improve as more key residues for specificity and activity are discovered.
Browse Plasmids: Single-Strand Break (Nick), Double-Strand Break (Cut)
Increasing Cas9 PAM flexibility
CRISPR systems designed for more precise edits require PAM sequences in very close proximity to the region to be edited, making the PAM sequence (NGG) limiting. This PAM is abundant throughout the human genome, but an NGG sequence may not be positioned correctly to target your desired genes for modification. Engineered SpCas9 enzymes with altered PAM specificities are available, known as PAM-flexible or PAMless Cas9s. SpCas9-derived variants with non-NGG PAM sequences include:
- xCas9 — NG, GAA, and GAT; increased nuclease fidelity
- SpCas9-NG — NG; increased in vitro activity
- SpG — NGN; increased nuclease activity
- SpRY — NRN/NYN, where R = A or G, Y = C or T
- SpRYc — SpRY PAMs plus NNG; a chimeric version of SpRY
Though these PAM-flexible Cas9s are useful for precision edits, they can be used for virtually any CRISPR application.
Cas9 Orthologs
While SpCas9 is the most commonly used Cas enzyme, additional Cas9 orthologs are available. SaCas9 (from Staphylococcus aureus) has a coding sequence ~1 kb shorter than SpCas9 while retaining the same basic function. Its size allows it to be efficiently packaged into adeno-associated viruses (AAVs).
Other natural Cas orthologs include Hsp1Cas9 (from Helicobacter sp. MIT 11-5569) and Nme2Cas9 (from Neisseria meningitidis), which are about the same size as SaCas9, with higher fidelity than SpCas9. FrCas9 (from Faecalibaculum rodentium) has increased fidelity, and recognizes NNTA, a PAM sequence often found in plants, expanding the targeting range for plant genome engineering.
Read more about Cas9 variants.
Anti-CRISPR
Prolonged Cas9 activity can increase the risk of off-target cutting or cytotoxicity, but naturally-occurring Acr (Anti-CRISPR) proteins can help control CRISPR activity. Acr proteins have evolved in phages and other mobile genetic elements to overcome the CRISPR systems that bacteria and archaea use to protect against these foreign nucleic acids. Acr family members inhibit CRISPR by a variety of mechanisms, including blocking crRNA loading or PAM recognition, inhibiting Cas9’s endonuclease domain, or inducing degradation of the Cas9 protein.
Most Acr proteins are specific to a particular type of Cas enzyme, such as Cas9 or Cas12a. These are identified in their names — AcrII proteins inhibit Cas9 (CRISPR Type II) systems, while AcrV inhibits Cas12a (CRISPR Type V) systems. There are rare cases where Acr can work with multiple types, such as AcrVA3.1, which inhibits both Cas12a and Cas3.
Similarly, some Acr proteins are specific to a particular species of CRISPR enzymes, such as AcrIIA7–10, which inhibits SpCas9. More commonly, Acrs inhibit multiple species of Cas enzymes, such as AcrIIA2 and AcrIIA4, isolated from Listeria monocytogenes, which can inhibit the CRISPR activity of both LmoCas9 and SpCas9.
Acr proteins can be used to control both when and where genome editing happens. CASANOVA, developed by Dominik Niopek’s lab, is a way to optogenetically control genome editing by fusing the anti-CRISPR protein AcrIIA4 with the photosensitive LOV2 domain. Several labs have also controlled Acr activity with endogenous cell-type specific miRNAs to restrict CRISPR editing only to the target tissue.
Browse Plasmids: Anti-CRISPR
Small Precision Edits
CRISPR can be used to make small, very precise edits of a handful of bases, or even a single base. These small precision edits can be done via HDR, base editing, and prime editing, and are often used to change an amino acid, turn a promoter on/off, or add small protein tags.
Precise Modifications Using Homology Directed Repair (HDR)
HDR generates specific genomic edits by copying from a DNA donor template. The donor template contains the desired sequence and is delivered to the cell along with the gRNA(s) and Cas enzyme. The donor DNA contains the desired edit as well as additional DNA matching the sequence immediately upstream and downstream of the target site (termed left and right homology arms). The length of each homology arm depends on the size of the change being introduced; larger insertions require longer homology arms.
Depending on the application, the donor DNA may be a single-stranded oligonucleotide, double-stranded linear DNA, or a double-stranded DNA (dsDNA) plasmid. Small single-stranded DNA (ssDNA) oligos often work well for small edits like a single-point mutation, while whole plasmid or linear dsDNA templates work better for large edits or insertions. When designing the donor template, incorporating mutations that prevent further Cas9 cleavage will improve the accuracy and efficiency of the desired edit. For example, you should alter the PAM site or target sequence in your donor DNA with a silent mutation that does not change the amino acid sequence.
HDR efficiency is generally low (< 10% of modified alleles). Selecting a target cut site as close as possible to the desired edit can increase efficiency. Synchronizing cells or modulating the cell cycle can help as HDR takes place during the S and G2 phases. Chemically or genetically inhibiting genes involved in NHEJ or upregulating those in the HDR pathway can also help. Additionally, constructs with Cas9 fused to proteins that modulate HDR or NHEJ components, like miCas9, Cas9-DN1S, or others, can increase HDR efficiency.
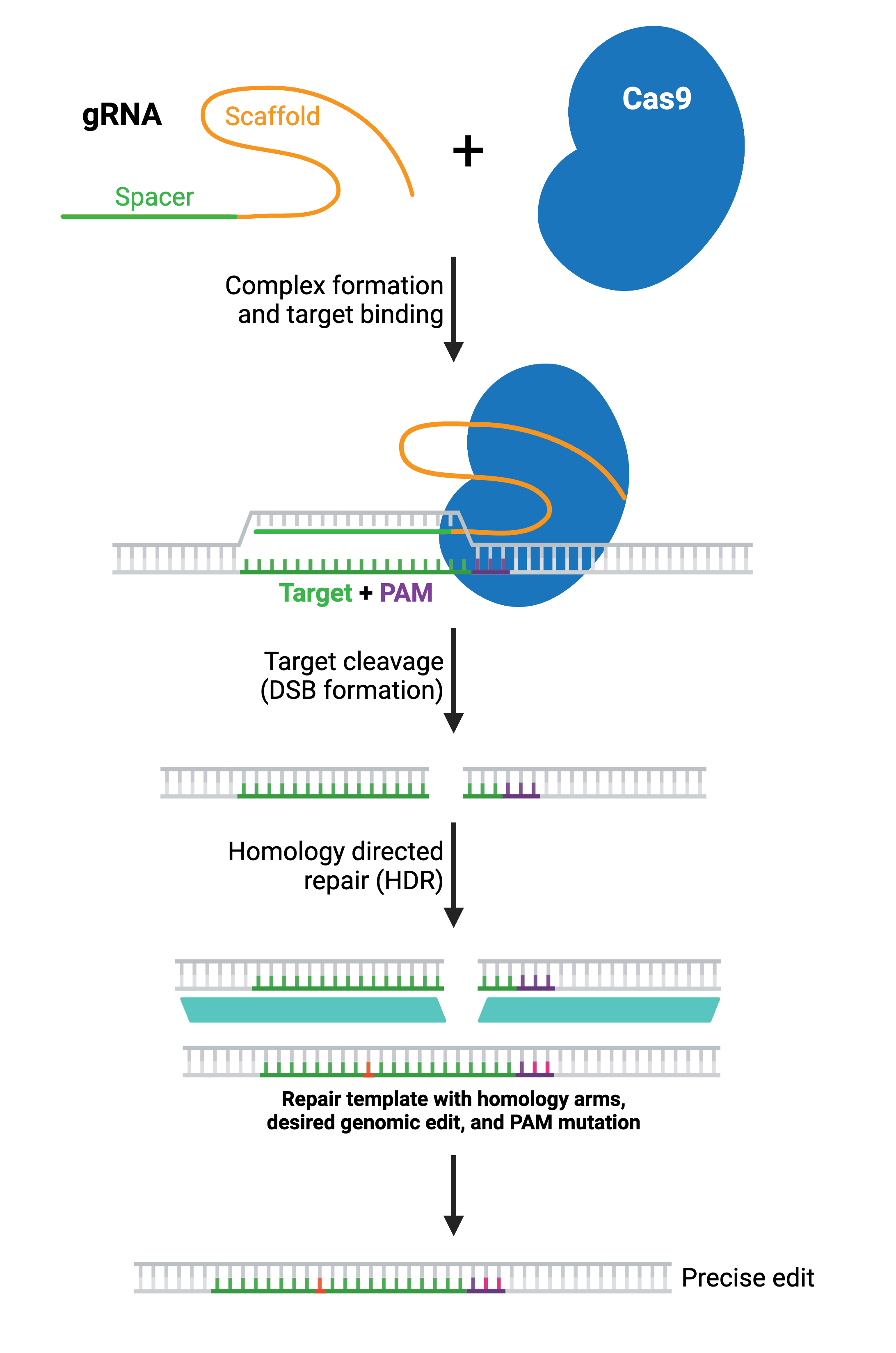
In most applications, a large portion of the Cas9-induced DSBs will still be repaired via NHEJ. The resulting population of cells will contain some combination of wild type alleles, NHEJ-repaired alleles, and the desired HDR-edited allele. Therefore, it is important to confirm the presence of the desired edit experimentally and to isolate clones containing the desired edit (see: Validate genetic modification).
Browse Plasmids: Double-Strand Break (Cut), Endogenous Tagging
CRISPR Base Editing
To overcome low HDR efficiency, researchers have developed base editors to specifically convert one base to another within a target locus of about 8 bp, specified by a gRNA. There are two main types of base edits:
- Base transitions — edit to same classification of nucleotide base (purine-to-purine or pyrimidine-to-pyrimidine)
- Base transversions — edit to different classification of nucleotide base (purine-to-pyrimidine and vice versa)
The first classes of base editors were cytosine base editors (pyrimidine-to-pyrimidine) and adenine base editors (purine-to-purine), which catalyze base transitions. Cytosine base editors are created by fusing Cas9 nickase (Cas9n) or dCas9 to a cytidine deaminase like APOBEC. Within the region of ssDNA created by Cas9 binding, the cytidine deaminase converts cytidine to uridine, which is read as thymidine during subsequent DNA repair, creating a C to T change. Modern base editors include several fused components that promote the intended repair pathway or inhibit unwanted ones and improve editing efficiency.
Likewise, adenine base editors have been engineered to convert adenosine to inosine, which is treated like guanosine by the cell, creating an A to G (or T to C) change. Adenosine DNA deaminases do not exist in nature, but have been created by directed evolution of the Escherichia coli TadA, a tRNA adenosine deaminase. Like cytosine base editors, the evolved TadA domain is fused to Cas9n or dCas9 to create the adenine base editor.
The TadA domain has also been further evolved to catalyze cytidine deamination instead of (or in addition to) adenosine deamination, producing cytosine base editors that are smaller and less likely to cause off-target mutations than those derived from naturally occurring cytidine deaminases.
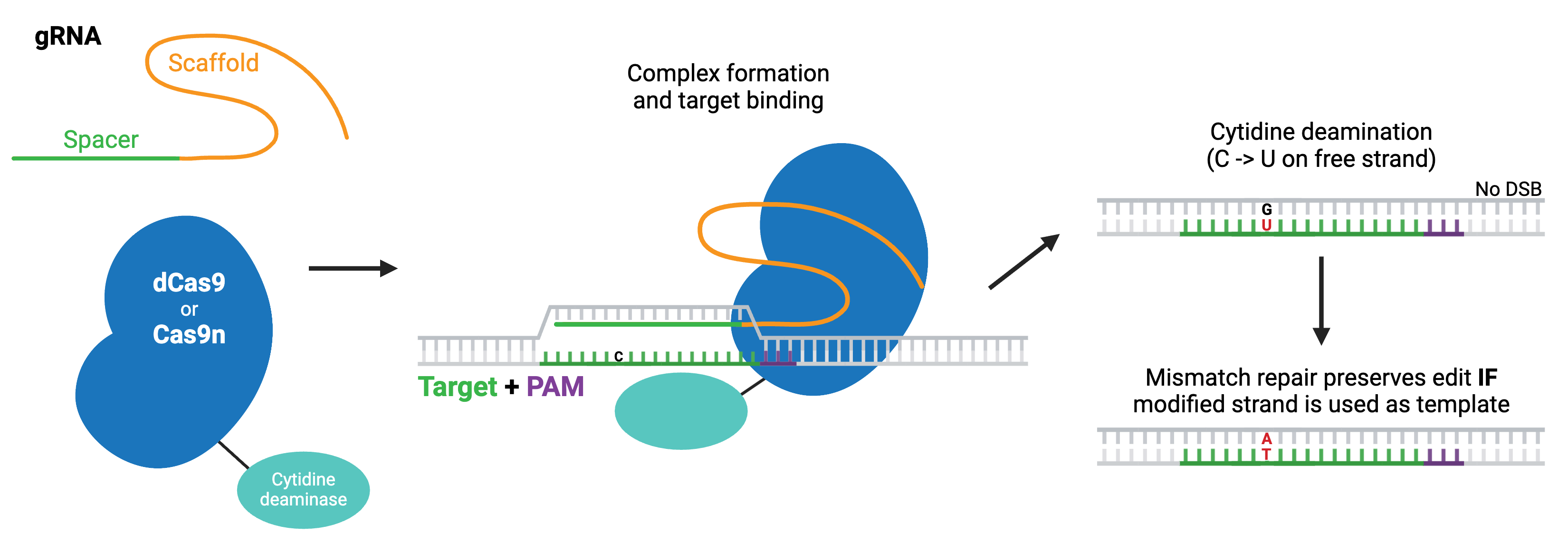
More recently, researchers have developed base editors that can catalyze base transversions. These editors are based on cytosine base editors and similarly work by first converting cytidine to uridine. However, they promote the base excision repair pathway, which fully removes the uridine from the DNA and replaces it with another base. In E. coli, this strategy yields C to A base edits, whereas in mammalian cells, it produces C to G edits.
Adenine transversion editors soon followed, using an enzyme evolved to excise inosine to access the same repair pathway and convert A to any other base. In principle, the ability to induce base transversions means that any base-to-base conversion is possible with base editing.
While many base editors are designed to work in a very narrow window proximal to the PAM sequence, some base editing systems create a wide spectrum of single-nucleotide variants (somatic hypermutation) in a larger editing window. These systems are well-suited to directed evolution applications. Examples of these base editing systems include targeted AID-mediated mutagenesis (TAM) and CRISPR-X, in which Cas9 is fused to activation-induced cytidine deaminase (AID).
Base editors are available with multiple Cas9 variants, including high fidelity Cas9s, Cas9s with various PAM requirements, and other Cas enzymes like Cas13. Specialized variants of the editing enzymes have been developed to reduce off-target mutations in DNA, RNA, or both, and are recommended in contexts where such mutations would be problematic.
Browse Plasmids: Base Edit
CRISPR Prime Editing
Prime editing is a “search and replace” gene editing method in which Moloney Murine Leukemia Virus Reverse Transcriptase (M-MLV RT) is fused to the C-terminus of Cas9 H840A nickase. The fusion enzyme can catalyze targeted insertions, deletions, and all possible base-to-base conversions using a prime editing guide RNA (pegRNA).
Like sgRNA, pegRNA directs the nickase to the target site by homology to a genomic DNA locus, where Cas9n nicks the non-target strand and generates a 3’ flap. However, the longer pegRNA also encodes a primer binding site (PBS) and the desired edits on a RT template. The PBS base pairs to the 3’ flap, and the desired edit is incorporated into the DNA by reverse transcription. The edited DNA strand displaces the unedited 5’ flap, and the resulting heteroduplex is resolved by the cell’s mismatch repair system. Alternatively, the edited 3’ flap may be excised and the target sequence will remain unchanged and available as a substrate for another round of prime editing.
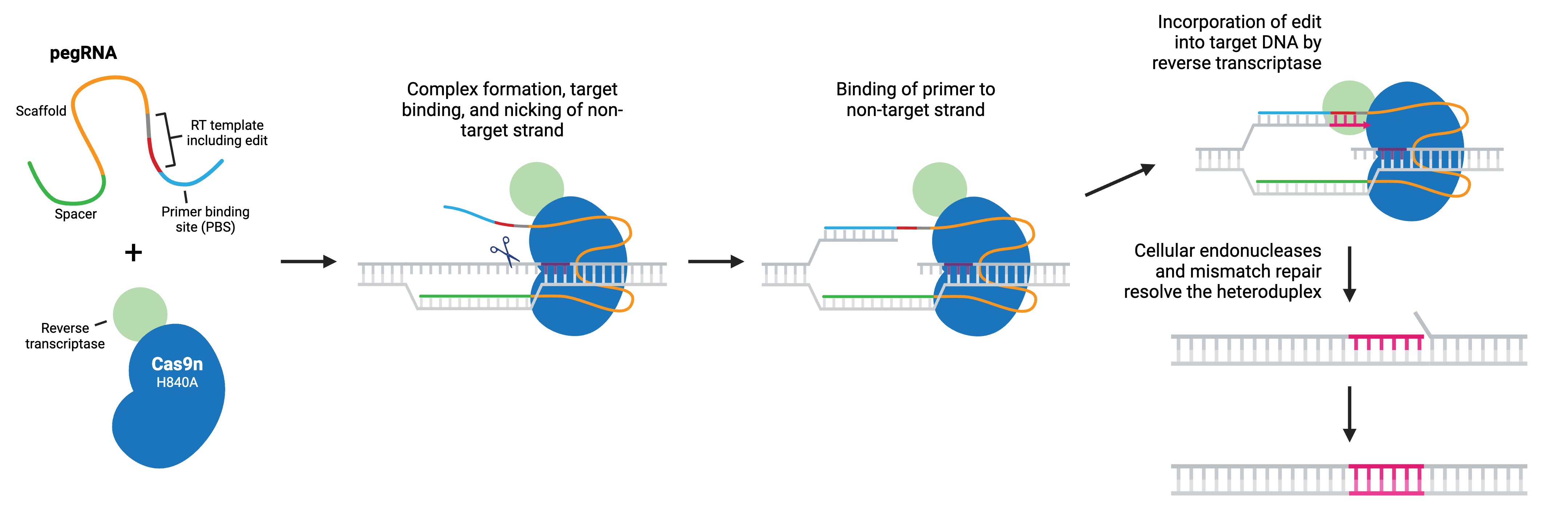
Popular prime editing (PE) tools include:
- PE2 — improved RT enzyme efficiency
- PE3 — increased efficiency; an additional sgRNA directs Cas9n to a nearby site, driving mismatch repair using the edited strand as a template
- PE4/5 — improved PE2/PE3 to inhibit mismatch repair to promote desired edit
- epegRNA — addition of a structured motif to enhance pegRNA stability
- PEmax — optimized PE architecture; increased synergy between PE enzymes and epegRNAs for enhanced editing
- PE7 — addition of RNA-binding protein to stabilize pegRNAs
- Twin PE — two PEs make complementary edits to opposite strands
Prime editing and base editing both offer high efficiency and low indel formation compared to HDR. Prime editing requires more optimization and typically has lower efficiency (< 50%) than base editing (> 90%), while also producing more indels. Prime editing is more flexible than base editing: it can encode edits involving any new sequence (point mutations, insertions, or deletions) under ~100 bp, which also increases the distance from a PAM accessible for editing. Base editors tend to change every target base within their editing window, while prime editing can specify a single mutation, even within a tract of identical bases.
Browse Plasmids: Prime Edit
Large Scale Edits
CRISPR can generate large-scale genetic changes, including moving entire genes to different locations, by combining other gene editing mechanisms, including transposons, integrases, and recombinases, with Cas enzymes.
CRISPR Transposases
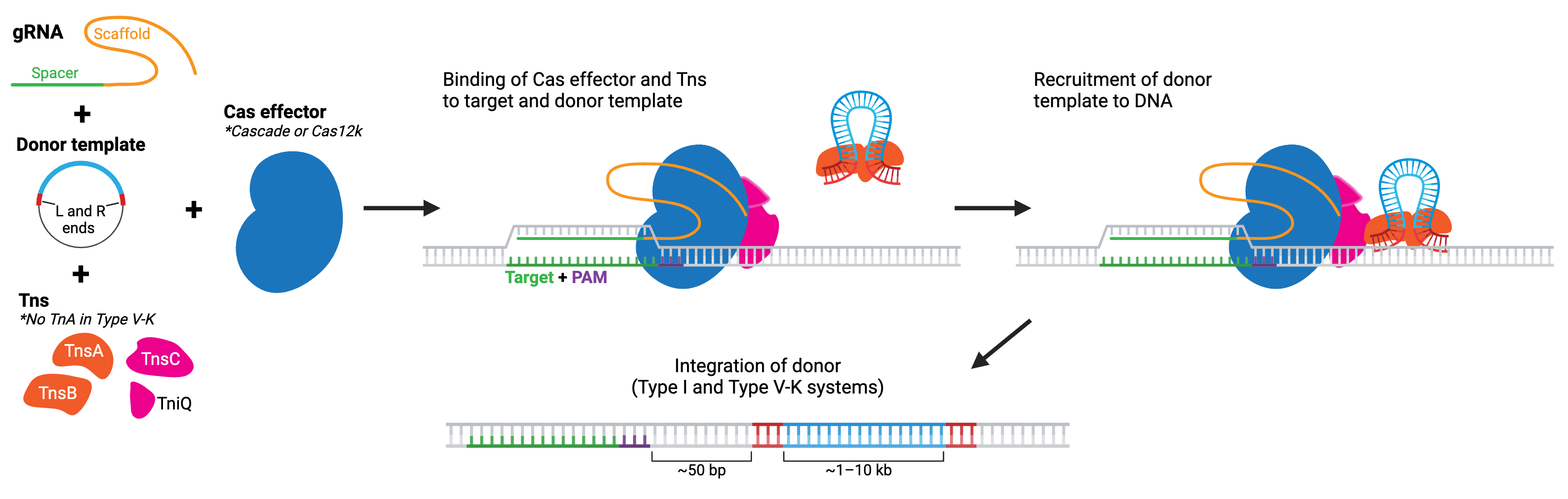
Transposon systems like piggyBac or Tn7 allow for large scale edits by integrating segments of DNA flanked by specific repeat elements into the genome. CRISPR-associated transposases (CASTs, developed by the Feng Zhang lab) combine the large-scale capacity of transposases with the precise DNA-targeting of CRISPR-Cas systems. You may also see them referred to as INTEGRATE (Insertion of Transposable Elements by Guide RNA-Assisted Targeting), developed by Samuel H. Sternberg’s lab. Many of these systems are based on naturally occurring CRISPR-associated Tn7-like transposases found in diverse species of microbes. However, synthetic dCas9-transposase fusions have also been developed.
The two main classes of CAST systems are type I and type V-K. Type I systems use a type I Cas effector, composed of several separate Cas enzymes that form a complex called Cascade (see Cas3 for more details), while type V-K systems use a type V-K Cas effector, Cas12k. No matter the system, the Cas effectors lack nuclease activity. Each system also includes Tn7-like transposase components. Type I (generally) includes tnsA, tnsB, tnsC, and tniQ. In comparison, type V-K includes just tnsB, tnsC, and tniQ. The CRISPR-Cas complex recruits the transposase proteins to the target location in the genome, where the transposase then carries out integration of donor DNA. Both types of CASTs have been found to achieve donor DNA integration for sequences > 10 kb!
Currently, these systems perform best in bacterial species, but newer versions engineered for eukaryotic cells are available and further improvements are actively in progress. Some type I CASTs can result in bidirectional insertions — meaning they may not be suitable for applications where orientation of the insert is important. However, type I-F CASTs favor insertion in the T-RL direction, with the right homology end closest to the target site. Type V-K CASTs appear to exhibit almost unidirectional insertions but are more prone to off-target insertions. And in the case of both types, the transposon end sequences are integrated along with the sequence, so the edit is not considered “scarless”.
Browse Plasmids: CRISPR transposases
CRISPR Recombinases and Integrases
Recombinases, and their subfamily integrases, are used for large scale edits. These proteins, like Cre recombinase or phage derived serine integrases, insert donor DNA at “landing sites” defined by relatively short sequences. Some have been paired with CRISPR systems to create useful tools for large scale edits.
PASSIGE (Prime-Editing-Assisted Site-Specific Integrase Gene Editing), developed by the David Liu lab, pairs prime editing with a large serine recombinase (LSR). A prime editor is used to install the LSR Bxb1’s landing site into the genome at a desired locus, then Bxb1 installs the donor sequence. Bxb1 versions named evoBxb1 or eeBxb1 are evolved versions of the recombinase with improved integration efficiencies in mammalian cells.
PASTE (Programmable Addition via Site-specific Targeting Elements), developed by the Abudayyeh-Gootenberg lab, also builds off prime editing. PASTE uses a CRISPR-Cas9n fused to both a reverse-transcriptase and a serine integrase (Bxb1), along with two gRNAs — one pegRNA to introduce the integrase landing site (attB) and the other to stimulate DNA repair. This can result in more efficient integration of donor DNA.
Genome-Wide Screens Using CRISPR
In a genetic screen, a population of cells with mutations in — or activation/repression of — a wide variety of genes is generated and used to identify the genetic perturbations (often called hits) that result in a desired phenotype. The ease of gRNA design and synthesis, as well as the ability to target almost any genomic locus, makes CRISPR an ideal genome engineering system for large-scale, forward genetic screening.
CRISPR can make highly specific, permanent genetic modifications to eliminate target gene function. It has also been used extensively to screen for novel genes that regulate known phenotypes, including resistances to chemotherapy drugs and toxins, cell viability, and tumor metastasis. One of the most popular methods for conducting genome-wide screens using CRISPR involves pooled lentiviral CRISPR libraries.
What Are Pooled Lentiviral CRISPR Libraries?
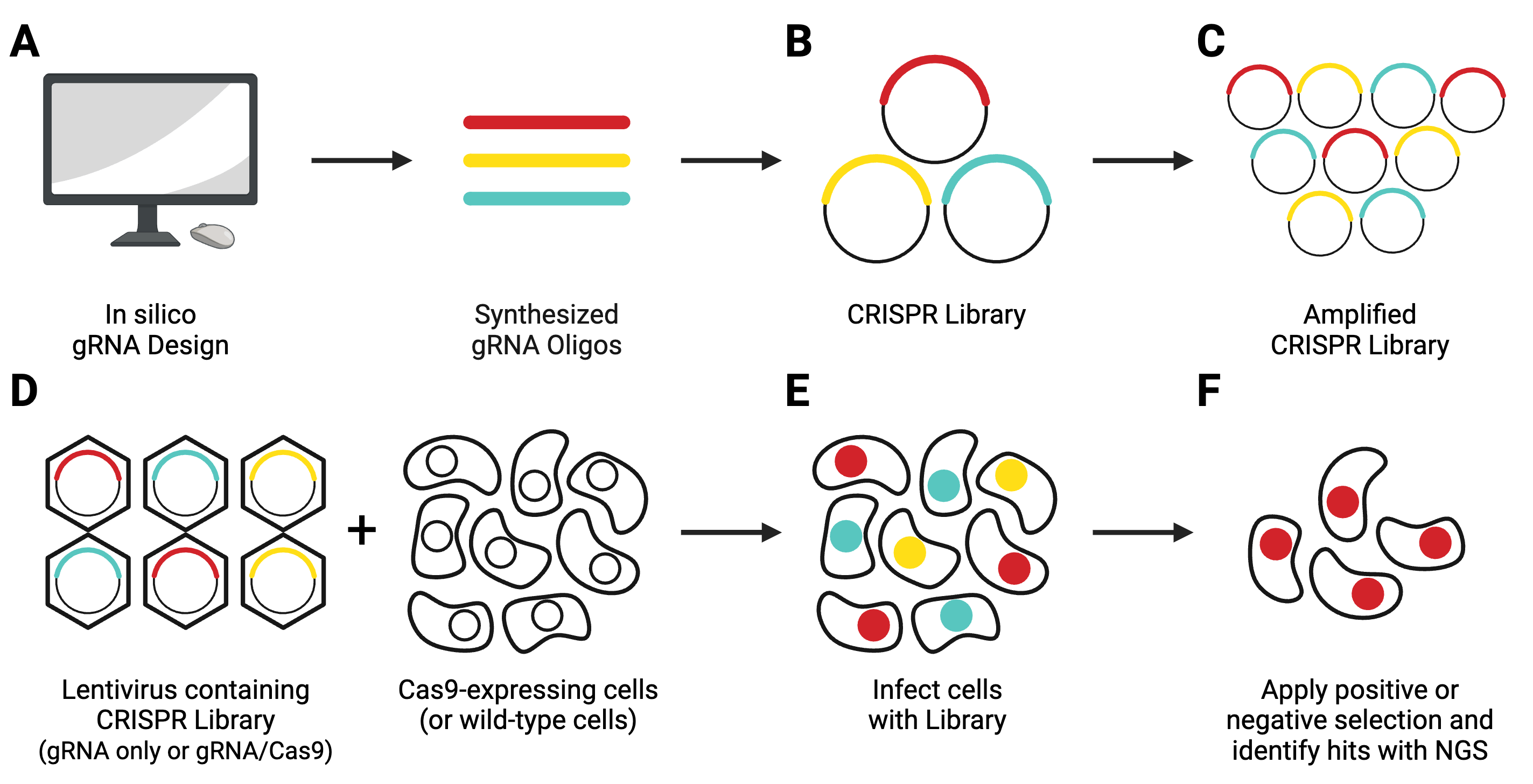
Pooled lentiviral CRISPR libraries are a heterogeneous population of lentiviral transfer vectors. In most cases, each plasmid contains an individual gRNA targeting a single gene in a given genome. Some more recent libraries use arrays of multiple gRNAs to target paralogs or lncRNAs.
Guide RNAs are designed in silico and synthesized (Figure 8A), then cloned in a pooled format into lentiviral transfer vectors (Figure 8B). Libraries come in 1-vector systems, in which Cas9 is included in the gRNA-containing plasmid, or 2-vector systems, in which Cas9 must be delivered separately. They can knockout, activate, or repress genes.
Each library typically contains ∼3–6 gRNAs per gene to ensure modification of every target gene, so CRISPR libraries contain thousands of unique gRNAs. gRNA design for CRISPR libraries is usually optimized to select for gRNAs with high on-target activity and low off-target activity. Knockout libraries often target 5′ constitutively expressed exons, while activation and repression libraries target promoter or enhancer regions. As each library is unique, be sure to check the library information and original publication to see if a library is suitable for your experiment.
Although lentiviral libraries are the most popular method for CRISPR screening, they are not suitable for all cell types or experiments. Mammalian CRISPR libraries have also been created in AAV backbones for in vivo experiments and in a retroviral backbone for delivery to cells that are poorly transduced by lentivirus. Non-mammalian CRISPR libraries are also available, such as those for yeast or Drosophila. Although CRISPR has been less widely used in bacteria due to technical challenges, bacterial CRISPR libraries have been developed for inhibition using dCas9.
How Do You Use a CRISPR Library?
CRISPR libraries from Addgene are available in two formats: as liquid DNA, or in select cases, as pre-made lentivirus.
DNA libraries are shipped at a concentration too low to be used in experiments and must be amplified before use (Figure 8C), following the amplification protocol specified by the depositing lab. After amplification, you’ll need to use next-generation sequencing (NGS) to verify that the composition of your amplified library matches that of the original library.
Next, you’ll need to generate lentivirus containing the entire library (Figure 8D). This lentivirus will then be used to transduce your experimental cell population (Figure 8E). In a 2-vector system, you’ll need to either co-infect with a Cas9-producing lentivirus or use a Cas9-expressing stable cell line. After applying your screening conditions to your transduced cell population, you will look for relevant hits using NGS technology.
For more detail on using CRISPR libraries for screens, see our pooled library guide and our blog post on lentiviral pooled libraries.
Browse Libraries: CRISPR Pooled Libraries
Cas9 Fusion Tools
dCas9 cannot cleave DNA, but it retains the ability to find specific DNA targets through a gRNA. Since dCas9 can be fused to other functional proteins to achieve specific outcomes, it has become a flexible tool to activate, repress, visualize, and isolate genes.
Activation or Repression of Target Genes
Targeting dCas9 to transcription start sites is sufficient to repress transcription by blocking initiation. More robust transcriptional repression (CRISPR interference, or CRISPRi) or activation (CRISPRa) of downstream target genes can be achieved by fusing dCas9 to transcriptional repressors or activators and targeting promoter regions. You might sometimes see these referred to together as CRISPRi/a.
dCas9-mediated gene activation or repression is reversible, as it does not permanently modify the genomic DNA. dCas9 has also been used for genome-wide screens to activate or repress gene expression in mice and human cells.
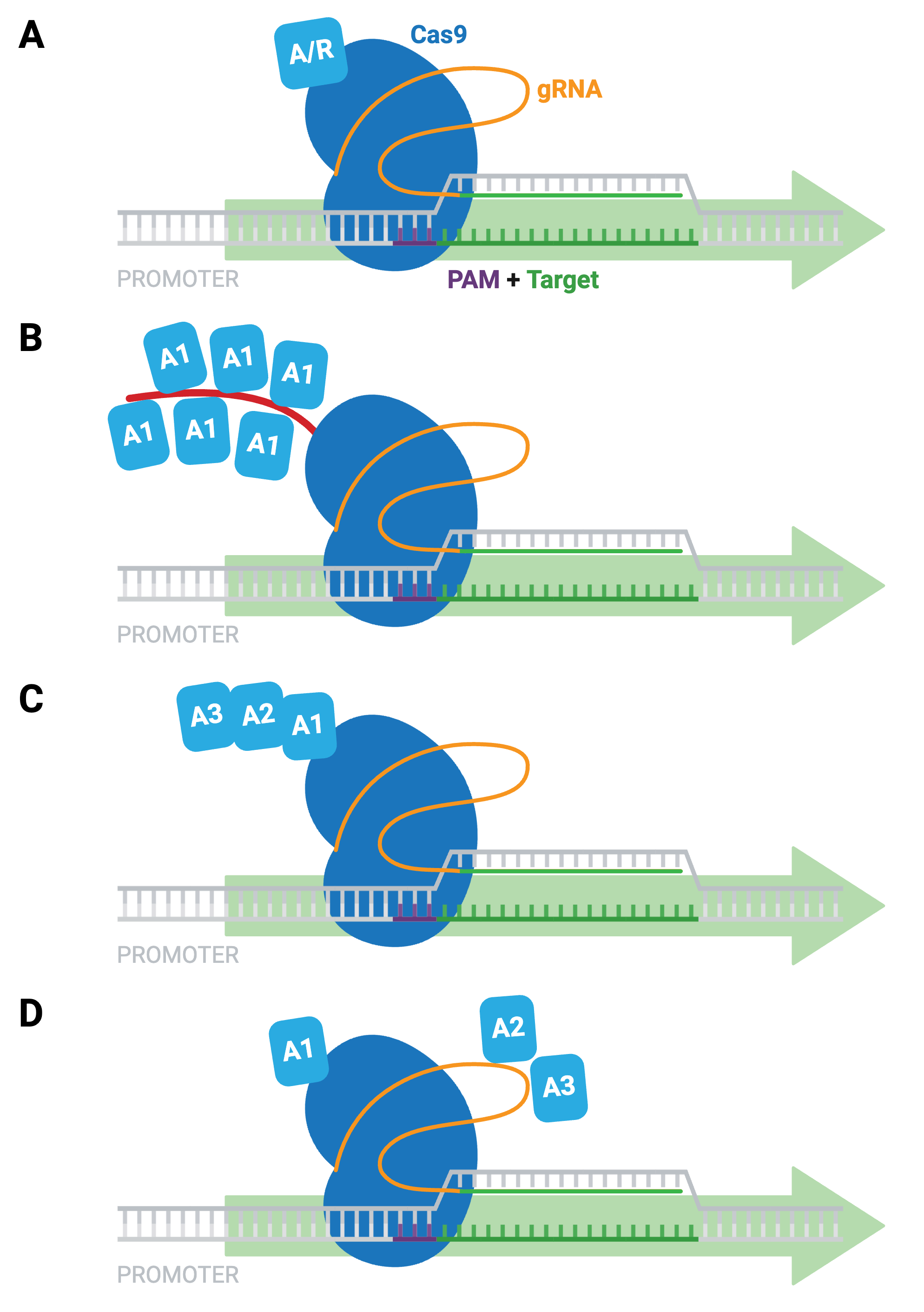
The simplest dCas9-based activators and repressors consist of dCas9 fused directly to a single transcriptional activator (e.g. VP64) or repressor (e.g. KRAB; Figure 9A). Other examples of activation systems to target genes in mammalian cells include:
- SunTag system — co-expression of epitope-tagged dCas9 and antibody-activator effector proteins; long-term imaging of proteins in living cells (Figure 9B)
- dCas9-VPR — dCas9 fused to several different activation domains in series; a single fusion protein, instead of a two-component system (Figure 9C)
- SAM activators — co-expression of dCas9-VP64 with a modified scaffold gRNA and additional RNA-binding helper activators; highest levels of single-gene activation (Figure 9D)
In bacteria, activating gene expression is more difficult because of the absence of effective gene activators when fused with dCas9. Recently, synthetic CRISPR-Cas gene activators have been developed for bacteria by using a scaffold RNA that contains the gRNA and an RNA hairpin to recruit activation proteins.
CRISPRi has also been adapted for diverse bacterial species using a modular system called Mobile-CRISPRi. In this system, CRISPRi is introduced into bacteria using conjugation and stably integrated into the chromosome.
Browse Plasmids: Activate, Repress/Interfere
Epigenetic Modifications Using CRISPR
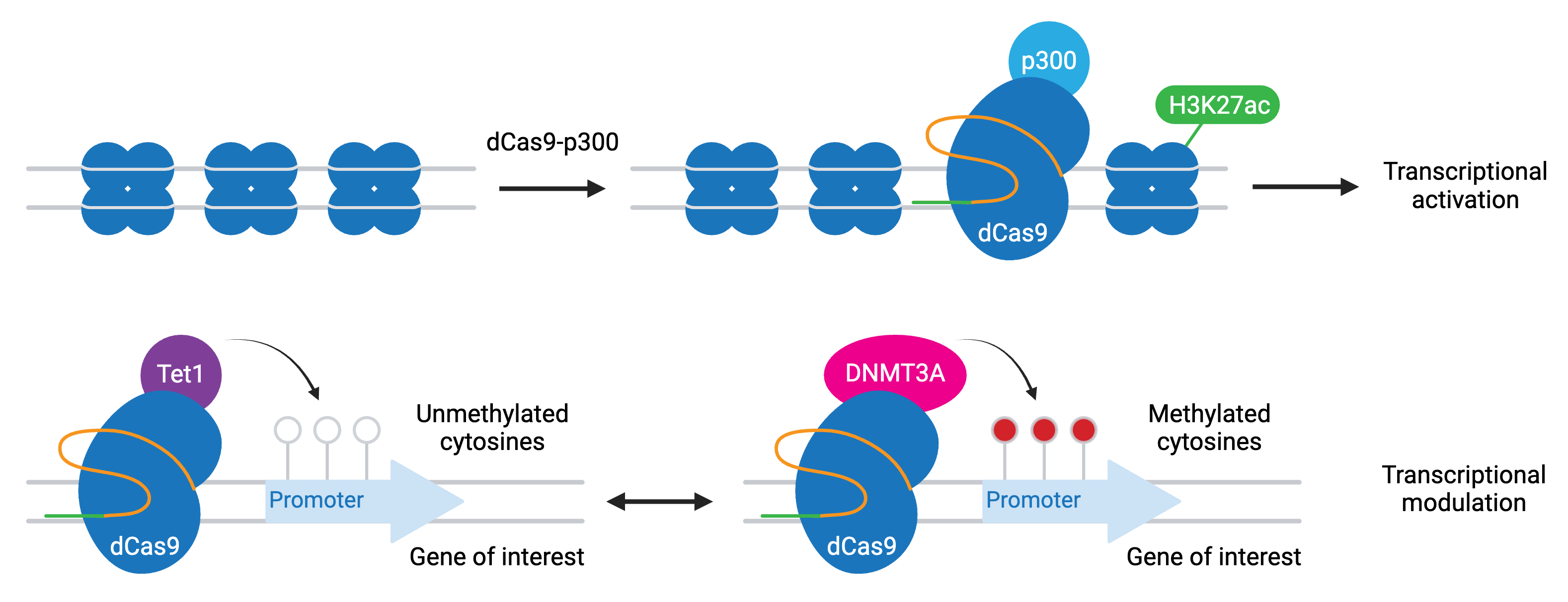
A variation of CRISPRi/a involves dCas9 fused to epigenetic modifiers. These programmable epigenome engineering tools alter gene expression by modifying the methylation state of cytosines in a gene’s promoter or by inducing histone acetylation or demethylation. The most common modifiers include:
- P300 — activation through increased H3K27 acetylation
- TET1 — activation through cytosine demethylation
- DNMT3A — repression through cytosine methylation
- MQ1 — repression through cytosine methylation with improved kinetics
- LSD1 — repression through targeted removal of H3K4 and H3K9 methylation
In certain cases, epigenetic modifiers may be a better choice than other activators/repressors due to their persistence and heritability. Not all modifications are stable across cell divisions, and some types of modifications are more frequently inherited by daughter cells. To maintain activation or repression, many CRISPR epigenetic tools require constitutive expression of the dCas9 fusion and/or expression of multiple fusions. The effects of CRISPRi/a may be reversible once the effector is inactivated or removed from the system.
CRISPRoff, developed by the Luke Gilbert and Jonathan Weissman labs, is an all-in-one dCas9 fusion with KRAB, DNMT3A, and DNMT3L. CRISPRoff maintains gene silencing across much of the human genome following transient expression of the dCas9 fusion, with the epigenetic modifications persisting through many generations and even cell differentiation. The system is reversible, with a companion CRISPRon to undo repression using a TET1-dCas9 fusion.
Browse Plasmids: Epigenetics
Visualizing Genomic Loci Using Fluorophores
CRISPR imaging has numerous advantages, including the simplicity of gRNA design, programmability for different genomic loci, and the capability to detect multiple targets simultaneously. CRISPR imaging can visualize chromatin and RNA dynamics in live cells.
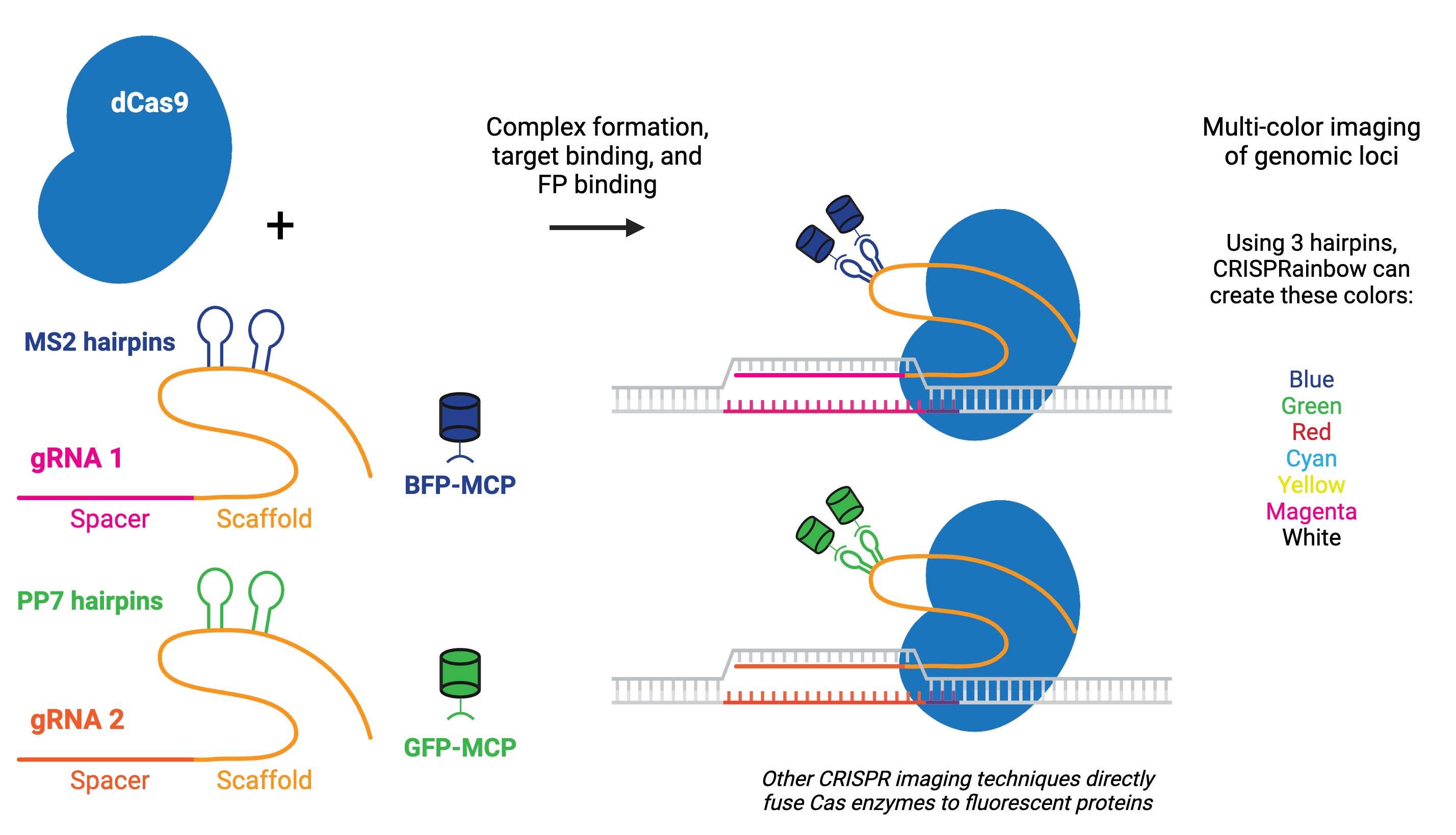
Labeling can be done by fusing catalytically inactive dCas9 to a fluorescent marker like green fluorescent protein (GFP), creating a customizable DNA or RNA label for fluorescence microscopy. RNA aptamers, like MS2 or PP7, can also be fused to the gRNA to recruit fluorescently-tagged RNA-binding proteins (RBPs).
Multicolor CRISPR imaging can be achieved by using orthogonal dCas9s (e.g. S. pyogenes dCas9 and S. aureus dCas9) tagged with different fluorescent proteins or by fusing the gRNAs to orthogonal protein-interacting RNA aptamers, such as in the CRISPRainbow kit. Using multiple copies of the aptamer improves stability and amplifies the signal for better imaging of genomic loci, as in the CRISPR-Sirius.
Fluorescent CRISPR systems have been used for dynamic tracking of repetitive and non-repetitive genomic loci, chromosome painting, and RNA tracking in living cells. Visualizing a specific genomic locus requires recruitment of many copies of labeled proteins to the site. Chromosome-specific repetitive loci can be efficiently visualized in living cells using a single gRNA that has multiple target sequences in close proximity. A non-repetitive genomic locus can also be labeled by co-delivering multiple gRNAs that tile the locus. Chromosome painting requires delivery of hundreds of gRNAs with target sites throughout the chromosome. Using modified dCas9, Csm, or Cas13 family proteins that target RNA instead of DNA enables tracking of RNA in live cells, including orthogonal RNA/DNA labeling in the same cell.
Browse Plasmids: Visualize
Purify Genomic Regions
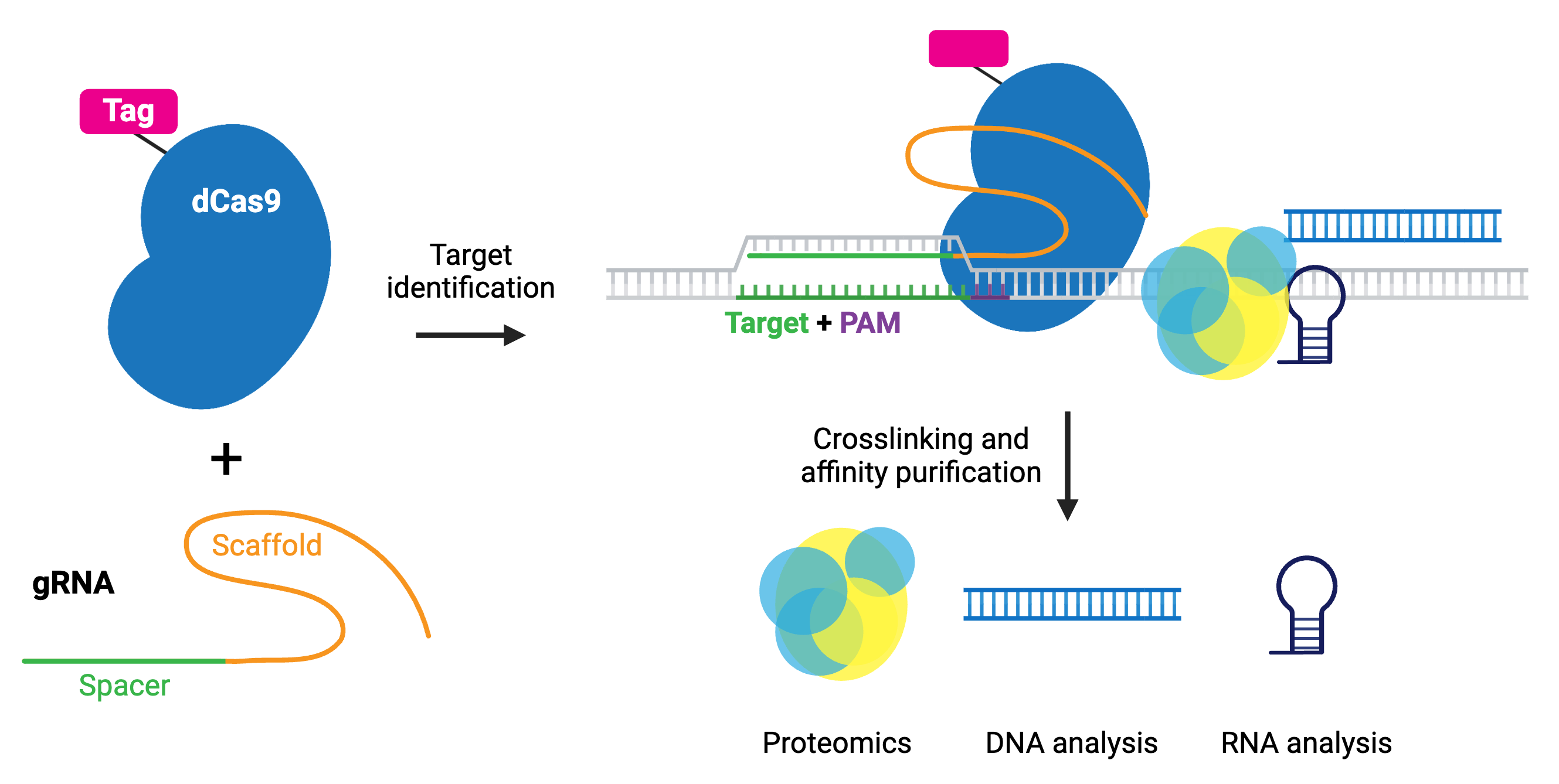
The enChIP (engineered DNA-binding molecule-mediated ChIP) system, developed by the Hodaka Fujii lab, pairs CRISPR with chromatin immunoprecipitation (ChIP) to allow purification of any genomic sequence specified by a particular gRNA. Epitope tag(s) are fused to dCas9 or gRNA for efficient purification of target DNA. The system can use common tags, like 3xFLAG-tag, PA, and biotin tags, or an anti-Cas9 antibody.
The CAPTURE system, developed by the Jian Xu lab, utilizes biotin tagging of dCas9 by fusing a biotin acceptor site to dCas9 and co-expressing BirA biotin ligase. Whatever the chosen epitope tag, the locus is subsequently isolated by affinity purification.
After purification, molecules associated with the locus can be identified by mass spectrometry (proteins), RNA-sequencing (RNAs), and NGS (other genomic regions). Compared to conventional methods for genomic purification, CRISPR-based purification methods are more straightforward and enable direct identification of molecules associated with a genomic region of interest in vivo.
Browse Plasmids: Purify
Cas9 Alternatives for CRISPR Genome Engineering
S. pyogenes Cas9 (SpCas9) is the most commonly used CRISPR endonuclease for genome engineering, but there are other Cas enzymes available. These enzymes have unique properties and can target different types of nucleic acids, including dsDNA (Cas9, Cas12), ssRNA (Cas13), or ssDNA (Cas14).
Efficient Editing and Multiplexing with Cas12a
One of the most popular Cas9 alternatives is Cas12a (Cpf1). There are many Cas12a orthologs, but the most common is AsCas12a (from Acidaminococcus sp.). The popularity can be attributed to Cas12a’s many benefits.
Cas12a exhibits increased efficiency with larger edits using HDR. Cas12a-mediated DNA cleavage creates DSBs with a short 3′ overhang, allowing for directional gene transfer, which may increase the efficiency of gene editing. Cas12a also has more condensed multiplexing capabilities, as it can process multiple gRNAs under a single promoter, and can cleave ssDNA non-specifically.
Cas12a also has an expanded targeting range to AT-rich regions, as it recognizes TTTV (V = A, C, or G) PAM sites. Engineered versions of Cas12a recognize different PAM variants, with one of the most popular being EnCas12a, an engineered version of AsCas12a that recognizes TYCV and TATV PAM sites.
The DETECTR platform, developed by the Jennifer Doudna lab, can identify specific strains of pathogens or genetic mutations. In DETECTR, Cas12a is targeted to a specific genomic location, such as a viral genome, and a ssDNA-fluorescently quenched reporter is added. As this reporter is degraded by Cas12a, it will release a quantifiable signal that indicates the presence of your target DNA sequence.
Browse Plasmids: Cas12a
RNA Targeting and Editing using Cas13
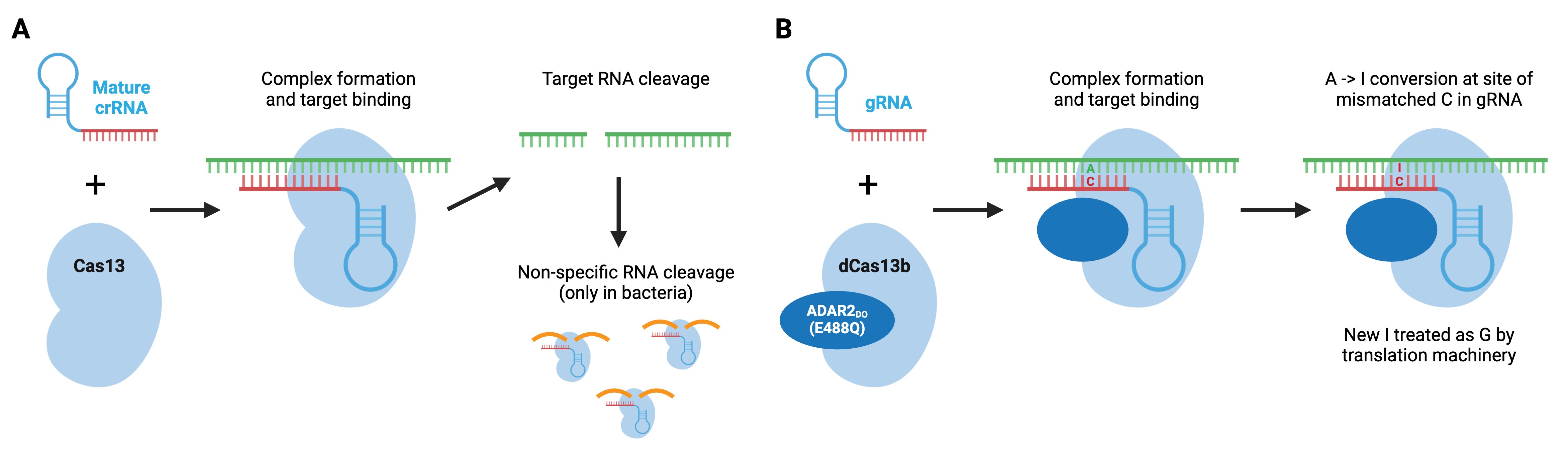
Cas13 is a type VI CRISPR enzyme that recognizes ssRNA. Type VI enzymes, which include Cas13a, Cas13b, and Cas13d, can process crRNA precursors to mature crRNAs. Upon ssRNA recognition by the crRNA, the target RNA is degraded. In bacteria, Cas13 enzymes can also cleave RNAs non-specifically after the initial crRNA-guided cleavage, which slows bacterial cell growth and may further protect bacteria from viral pathogens. This promiscuity is unique to bacterial cells and does not occur in mammalian cells.
SHERLOCK, developed by the Feng Zhang lab, is a Cas13a-based molecular detection platform. Based on this non-specific cleavage, a Cas13a-based molecular detection platform, termed SHERLOCK, can identify target sequences using a fluorescent reporter. In SHERLOCK, a quenched ssRNA fluorescent reporter is added to the reaction with the Cas13a and gRNA. When Cas13a binds to the target sequence, it promiscuously cleaves the reporter as well, and a quantifiable signal is produced, indicating the presence of that specific target. SHERLOCK (and SHERLOCKv2) allows for greater amplification and quantification and can be used on lateral flow strips. Other Cas enzymes can be added to SHERLOCKv2 to detect multiple targets. For example, Cas12 can be added to detect the presence of infectious organisms (like SARS-CoV-2) and genetic mutations.
Similar to Cas9 and Cas12, Cas13 can be converted to an RNA-binding protein through mutations in its catalytic domain. REPAIR, developed by the Feng Zhang lab, used a hyperactive adenosine deaminase that acts on RNA, ADAR2(E488Q) fused to catalytically dead Cas13b to create a programmable RNA base editor that converts adenosine to inosine in RNA. Since inosine is functionally equivalent to guanosine, the result is an A to G change in RNA. The catalytically inactive Cas13b ortholog from Prevotella sp., dPspCas13b, does not require a specific sequence adjacent to the RNA target, making this a very flexible editing system. Additional editors have been developed, including editors based on a second ADAR variant that displays improved specificity, and editors carrying an ADAR truncation that retain RNA editing capabilities but are small enough to be packaged in AAV particles.
Cas13 fusions have also been used in in vivo RNA imaging or to control alternative splicing.
Browse Plasmids: RNA Targeting, RNA Editing
Degrading DNA with Cas3
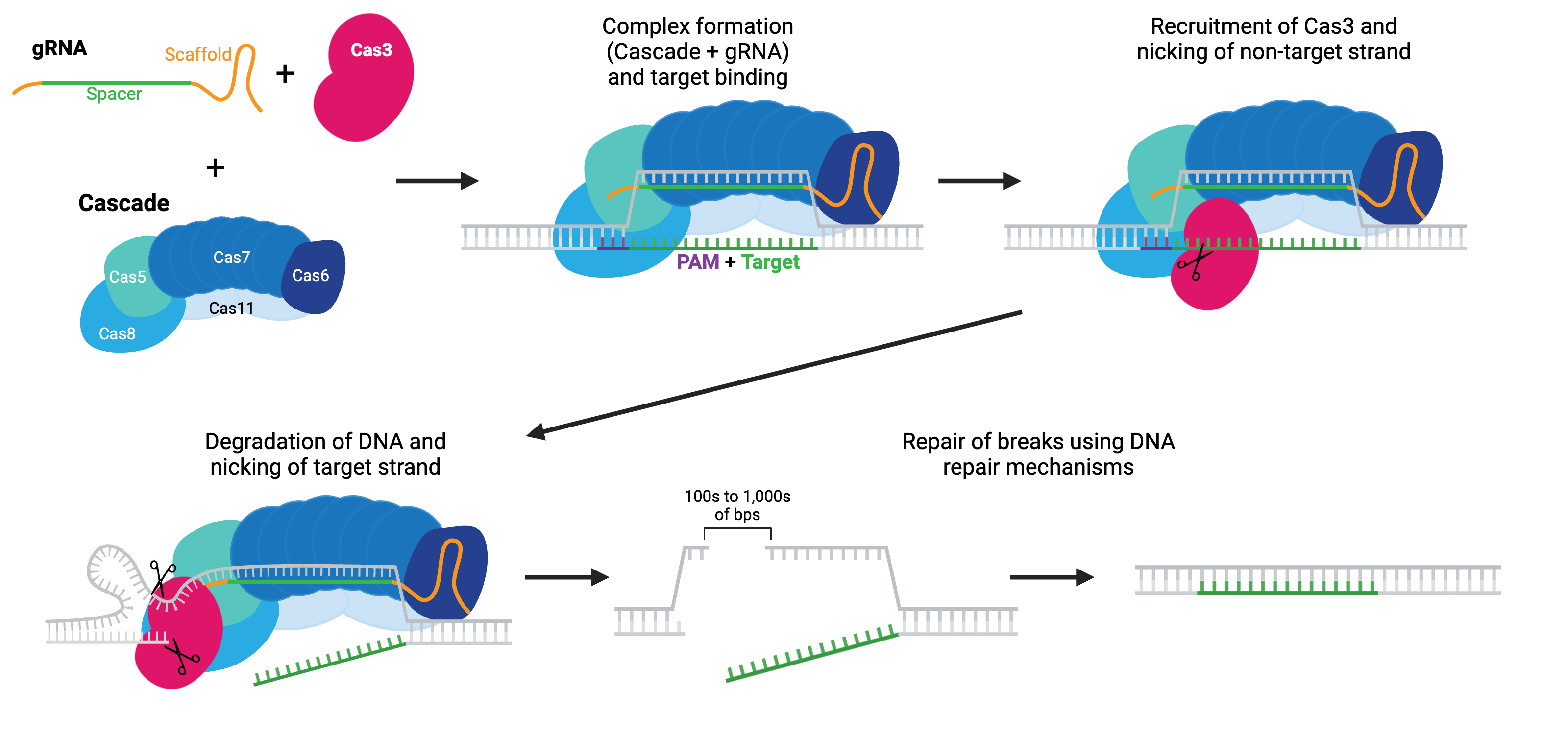
Rather than initiating DNA cleavage to form a break, Cas3 initiates single-strand DNA degradation. This degradation can continue for many kilobases in one or both directions from the target region, effectively turning Cas3 into a DNA “shredder” system that can cause large genomic deletions. The boundaries of these deletions can be defined, either with HDR repair templates or with anti-CRISPR proteins. Large deletions generated by Cas3 have fewer off-target alterations, such as small mutations or inversions, than do large deletions generated by Cas9 systems.
Cas3 must be paired with the Cascade (CRISPR-associated complex for antiviral defense) complex to initiate DNA degradation. Cascade is made of five proteins: Cas8 (Cse1), Cas11 (Cse2), Cas7, Cas5 and Cas6e. As the name implies, this system is harnessed for antiviral mechanisms in bacteria (E. coli). Like the other Cas enzymes, the Cascade-Cas3 system has been co-opted for use in eukaryotic cells. The Cascade complex is recruited to target DNA using a gRNA and recruits Cas3. Cas3 nicks the non-targeting strand and starts degrading the single-stranded DNA, unraveling DNA as it goes using its helicase properties, often called a reeling mechanism. The targeted strand is cleaved non-specifically by Cas3 along the way. Cas3 initiates a final DSB, either due to a defined boundary or running into other proteins bound to DNA, and the break can be repaired by the cell’s endogenous repair mechanisms.
Browse Plasmids: Cascade-Cas3
Want more information on the wide variety of Cas enzymes? CasPEDIA is an encyclopedia of Class 2 CRISPR systems with wiki entries describing enzyme activity, experimental considerations, and more. CasPEDIA is a community resource created and maintained by the Jennifer Doudna lab at the Innovative Genomics Institute, University of California, Berkeley.
Resources
Figures
All figures were created using BioRender.com (Link opens in a new window).
Addgene Website & Blog References
Website
Blog
Cas Enzymes and PAM Sequences
| Species/Variant of Cas9 | PAM Sequence* |
|---|---|
| Streptococcus pyogenes (SP); SpCas9 | 3' NGG |
| SpCas9 D1135E variant | 3' NGG (reduced NAG binding) |
| SpCas9 VRER variant | 3' NGCG |
| SpCas9 EQR variant | 3' NGAG |
| SpCas9 VQR variant | 3' NGAN or NGNG |
| xCas9 | 3' NG, GAA, or GAT |
| SpCas9-NG | 3' NG |
| Staphylococcus aureus (SA); SaCas9 | 3' NNGRRT or NNGRR(N) |
| Acidaminococcus sp. (AsCpf1) and Lachnospiraceae bacterium (LbCpf1) | 5' TTTV |
| AsCpf1 RR variant | 5' TYCV |
| LbCpf1 RR variant | 5' TYCV |
| AsCpf1 RVR variant | 5' TATV |
| Campylobacter jejuni (CJ) | 3' NNNNRYAC |
| Neisseria meningitidis (NM) | 3' NNNNGATT |
| Streptococcus thermophilus (ST) | 3' NNAGAAW |
| Treponema denticola (TD) | 3' NAAAAC |
| Additional Cas9s from various species | PAM sequence may not be characterized |
*In the table above, 3' and 5' indicate on which end of targeted sequence the PAM is located. The majority of the CRISPR plasmids in Addgene’s collection are from S. pyogenes unless otherwise noted.
Glossary
| Term | Definition |
|---|---|
| Base editor | Fusion of a Cas protein to a deaminase that enables direct base conversion in RNA or DNA without a DNA double-strand break |
| Cas | CRISPR Associated Protein, includes nucleases like Cas9 and Cas12a (also known as Cpf1) |
| Cascade | CRISPR-associated complex for antiviral defense; a complex of multiple Cas enzymes including Cas8, Cas11, Cas7, Cas5, and Cas6e. Most often used with Cas3 and CRISPR transposases |
| Cas9n | Cas9 nickase; has one active and one inactive nuclease domain to generate DNA nicks by cutting only one strand |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeat, a bacterial genomic region used in pathogen defense |
| CRISPRa | CRISPR Activation; using a dCas9 or dCas9-activator with a gRNA to increase transcription of a target gene |
| CRISPRi | CRISPR Interference; using a dCas9 or dCas9-repressor with a gRNA to repress/decrease transcription of a target gene |
| Cut | A DNA double-strand break; the wild type function of most Cas nucleases |
| dCas9 | Nuclease dead Cas9, an enzymatically inactive form of Cas9; can bind, but cannot cleave DNA |
| DSB | Double-Strand Break, a break in both strands of DNA |
| Dual nickase/Double nick | A method to decrease off-target effects by using a single Cas9 nickase and 2 different gRNAs, which bind in close proximity on opposite strands of the DNA, to create a DSB |
| enChIP | Engineered DNA-binding molecule-mediated ChIP, using a tagged-dCas9+gRNA to purify specific genomic regions to identify molecules associated with the genomic regions |
| Genetic modification or manipulation | Any genetic perturbation, including genetic knockout, gene activation, or gene repression |
| gRNA | Guide RNA, a synthetic fusion of the endogenous bacterial crRNA and tracrRNA that provides both targeting specificity and scaffolding/binding ability for Cas9 nuclease. This synthetic fusion does not exist in nature and is also commonly referred to as an sgRNA. |
| gRNA scaffold sequence | The sequence within the gRNA that is responsible for Cas9 binding, it does not include the 20 bp spacer/targeting sequence that is used to guide Cas9 to target DNA |
| gRNA targeting sequence | The 20 nucleotides that precede the PAM sequence in the genomic DNA. This sequence is cloned into a gRNA expression plasmid but does NOT include the PAM sequence or the gRNA scaffold sequence |
| HDR | Homology Directed Repair, a DNA repair mechanism that uses a template to repair DNA nicks or DSBs |
| Indel | Insertion/deletion, a type of mutation that can result in the disruption of a gene by shifting the ORF and/or creating premature stop codons |
| NHEJ | Non-Homologous End Joining; A DNA repair mechanism that often introduces indels |
| Nick | A break in only one strand of dsDNA |
| Nickase | Cas9 with one of the two nuclease domains inactivated. This enzyme is capable of cleaving only one strand of target dsDNA. |
| Off-target effects or off-target activity | Cas9 cleavage at undesired locations due to gRNA targeting sequence with sufficient homology to recruit Cas9 to unintended genomic locations |
| On-target activity | Cas9 cleavage at a desired location specified by a gRNA target sequence |
| ORF | Open Reading Frame; The translated codons that make up a gene |
| PAM | Protospacer Adjacent Motif; Sequence adjacent to the target sequence that is necessary for Cas enzymes to bind target DNA |
| PCR | Polymerase Chain Reaction; Used to amplify a specific sequence of DNA |
| pegRNA | Prime Edit gRNA; a specialized gRNA containing an additional primer binding site and reverse transcription template with the desired edit |
| Prime editor | Fusion of a Cas9 H840A nickase and M-MLV RT (Moloney Murine Leukemia Virus Reverse Transcriptase) for "search and replace" editing |
| Target locus | Genomic target of the gRNA. The sequence includes the unique ~20 bp target specified by the gRNA plus the genomic PAM sequence |
Publications (by topic)
CRISPR basics
Anders, C., Niewoehner, O., Duerst, A., & Jinek, M. (2014). Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature, 513(7519), 569–573. https://doi.org/10.1038/nature13579 PMID: 25079318
Bondy-Denomy, J., Pawluk, A., Maxwell, K. L., & Davidson, A. R. (2013). Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature, 493(7432), 429–432. https://doi.org/10.1038/nature11723 PMID: 23242138
Bondy-Denomy, J., Garcia, B., Strum, S., Du, M., Rollins, M. F., Hidalgo-Reyes, Y., Wiedenheft, B., Maxwell, K. L., & Davidson, A. R. (2015). Multiple mechanisms for CRISPR–Cas inhibition by anti-CRISPR proteins. Nature, 526(7571), 136–139. https://doi.org/10.1038/nature15254 PMID: 26416740
Bravo, J. P. K., Liu, M., Hibshman, G. N., Dangerfield, T. L., Jung, K., McCool, R. S., Johnson, K. A., & Taylor, D. W. (2022). Structural basis for mismatch surveillance by CRISPR–Cas9. Nature, 603(7900), 343–347. https://doi.org/10.1038/s41586-022-04470-1 PMID: 35236982
Bubeck, F., Hoffmann, M. D., Harteveld, Z., Aschenbrenner, S., Bietz, A., Waldhauer, M. C., Börner, K., Fakhiri, J., Schmelas, C., Dietz, L., Grimm, D., Correia, B. E., Eils, R., & Niopek, D. (2018). Engineered anti-CRISPR proteins for optogenetic control of CRISPR–Cas9. Nature Methods, 15(11), 924–927. https://doi.org/10.1038/s41592-018-0178-9 PMID: 30377362
Casini, A., Olivieri, M., Petris, G., Montagna, C., Reginato, G., Maule, G., Lorenzin, F., Prandi, D., Romanel, A., Demichelis, F., Inga, A., & Cereseto, A. (2018). A highly specific SpCas9 variant is identified by in vivo screening in yeast. Nature Biotechnology, 36(3), 265–271. https://doi.org/10.1038/nbt.4066 PMID: 29431739
Chen, J. S., Dagdas, Y. S., Kleinstiver, B. P., Welch, M. M., Sousa, A. A., Harrington, L. B., Sternberg, S. H., Joung, J. K., Yildiz, A., & Doudna, J. A. (2017). Enhanced proofreading governs CRISPR–Cas9 targeting accuracy. Nature, 550(7676), 407–410. https://doi.org/10.1038/nature24268 PMID: 28931002
Cheng, A. W., Wang, H., Yang, H., Shi, L., Katz, Y., Theunissen, T. W., Rangarajan, S., Shivalila, C. S., Dadon, D. B., & Jaenisch, R. (2013). Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Research, 23(10), 1163–1171. https://doi.org/10.1038/cr.2013.122 PMID: 23979020
Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., Hsu, P. D., Wu, X., Jiang, W., Marraffini, L. A., & Zhang, F. (2013). Multiplex genome engineering using CRISPR/CAS systems. Science, 339(6121), 819–823. https://doi.org/10.1126/science.1231143 PMID: 23287718
Cui, Z., Tian, R., Huang, Z., Jin, Z., Li, L., Liu, J., Huang, Z., Xie, H., Liu, D., Mo, H., Zhou, R., Lang, B., Meng, B., Weng, H., & Hu, Z. (2022). FrCas9 is a CRISPR/Cas9 system with high editing efficiency and fidelity. Nature Communications, 13(1), 1425. https://doi.org/10.1038/s41467-022-29089-8 PMID: 35301321
Edraki, A., Mir, A., Ibraheim, R., Gainetdinov, I., Yoon, Y., Song, C., Cao, Y., Gallant, J., Xue, W., Rivera-Pérez, J. A., & Sontheimer, E. J. (2019). A Compact, High-Accuracy Cas9 with a Dinucleotide PAM for In Vivo Genome Editing. Molecular Cell, 73(4), 714-726.e4. https://doi.org/10.1016/j.molcel.2018.12.003 PMID: 30581144
Fu, Y., Foden, J. A., Khayter, C., Maeder, M. L., Reyon, D., Joung, J. K., & Sander, J. D. (2013). High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nature Biotechnology, 31(9), 822–826. https://doi.org/10.1038/nbt.2623 PMID: 23792628
Fu, Y., Sander, J. D., Reyon, D., Cascio, V. M., & Joung, J. K. (2014). Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nature Biotechnology, 32(3), 279–284. https://doi.org/10.1038/nbt.2808 PMID: 24463574
Gao, S., Wang, Y., Qi, T., Wei, J., Hu, Z., Liu, J., Sun, S., Liu, H., & Wang, Y. (2023). Genome editing with natural and engineered CjCas9 orthologs. Molecular Therapy, 31(4), 1177–1187. https://doi.org/10.1016/j.ymthe.2023.01.029 PMID: 36733251
Harrington, L. B., Doxzen, K. W., Ma, E., Liu, J., Knott, G. J., Edraki, A., Garcia, B., Amrani, N., Chen, J. S., Cofsky, J. C., Kranzusch, P. J., Sontheimer, E. J., Davidson, A. R., Maxwell, K. L., & Doudna, J. A. (2017). A Broad-Spectrum inhibitor of CRISPR-Cas9. Cell, 170(6), 1224-1233.e15. https://doi.org/10.1016/j.cell.2017.07.037 PMID: 28844692
Hibshman, G. N., Bravo, J. P. K., Hooper, M. M., Dangerfield, T. L., Zhang, H., Finkelstein, I. J., Johnson, K. A., & Taylor, D. W. (2024). Unraveling the mechanisms of PAMless DNA interrogation by SpRY-Cas9. Nature Communications, 15(1), 3663. https://doi.org/10.1038/s41467-024-47830-3 PMID: 38688943
Hsu, P. D., Scott, D. A., Weinstein, J. A., Ran, F. A., Konermann, S., Agarwala, V., Li, Y., Fine, E. J., Wu, X., Shalem, O., Cradick, T. J., Marraffini, L. A., Bao, G., & Zhang, F. (2013). DNA targeting specificity of RNA-guided Cas9 nucleases. Nature Biotechnology, 31(9), 827–832. https://doi.org/10.1038/nbt.2647 PMID: 23873081
Hu, J. H., Miller, S. M., Geurts, M. H., Tang, W., Chen, L., Sun, N., Zeina, C. M., Gao, X., Rees, H. A., Lin, Z., & Liu, D. R. (2018). Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature, 556(7699), 57–63. https://doi.org/10.1038/nature26155 PMID: 29512652
Jinek, M., Chylinski, K., Fonfara, I., Hauer, M., Doudna, J. A., & Charpentier, E. (2012). A programmable Dual-RNA–Guided DNA endonuclease in adaptive bacterial immunity. Science, 337(6096), 816–821. https://doi.org/10.1126/science.1225829 PMID: 22745249
Kabadi, A. M., Ousterout, D. G., Hilton, I. B., & Gersbach, C. A. (2014). Multiplex CRISPR/Cas9-based genome engineering from a single lentiviral vector. Nucleic Acids Research, 42(19), e147. https://doi.org/10.1093/nar/gku749 PMID: 25122746
Kleinstiver, B. P., Prew, M. S., Tsai, S. Q., Topkar, V. V., Nguyen, N. T., Zheng, Z., Gonzales, A. P. W., Li, Z., Peterson, R. T., Yeh, J. J., Aryee, M. J., & Joung, J. K. (2015). Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature, 523(7561), 481–485. https://doi.org/10.1038/nature14592 PMID: 26098369
Kleinstiver, B. P., Pattanayak, V., Prew, M. S., Tsai, S. Q., Nguyen, N. T., Zheng, Z., & Joung, J. K. (2016). High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature, 529(7587), 490–495. https://doi.org/10.1038/nature16526 PMID: 26735016
Lee, J. K., Jeong, E., Lee, J., Jung, M., Shin, E., Kim, Y., Lee, K., Jung, I., Kim, D., Kim, S., & Kim, J. (2018). Directed evolution of CRISPR-Cas9 to increase its specificity. Nature Communications, 9(1). https://doi.org/10.1038/s41467-018-05477-x PMID: 30082838
Maddalo, D., Manchado, E., Concepcion, C. P., Bonetti, C., Vidigal, J. A., Han, Y., Ogrodowski, P., Crippa, A., Rekhtman, N., De Stanchina, E., Lowe, S. W., & Ventura, A. (2014). In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature, 516(7531), 423–427. https://doi.org/10.1038/nature13902 PMID: 25337876
Mali, P., Yang, L., Esvelt, K. M., Aach, J., Guell, M., DiCarlo, J. E., Norville, J. E., & Church, G. M. (2013). RNA-Guided Human Genome Engineering via CAS9. Science, 339(6121), 823–826. https://doi.org/10.1126/science.1232033 PMID: 23287722
Makarova, K. S., Wolf, Y. I., Alkhnbashi, O. S., Costa, F., Shah, S. A., Saunders, S. J., Barrangou, R., Brouns, S. J. J., Charpentier, E., Haft, D. H., Horvath, P., Moineau, S., Mojica, F. J. M., Terns, R. M., Terns, M. P., White, M. F., Yakunin, A. F., Garrett, R. A., Van Der Oost, J., . . . Koonin, E. V. (2015). An updated evolutionary classification of CRISPR–Cas systems. Nature Reviews Microbiology, 13(11), 722–736. https://doi.org/10.1038/nrmicro3569 PMID: 26411297
Marino, N. D., Pinilla-Redondo, R., Csörgő, B., & Bondy-Denomy, J. (2020). Anti-CRISPR protein applications: natural brakes for CRISPR-Cas technologies. Nature Methods, 17(5), 471–479. https://doi.org/10.1038/s41592-020-0771-6 PMID: 32203383
Nishimasu, H., Ran, F. A., Hsu, P. D., Konermann, S., Shehata, S. I., Dohmae, N., Ishitani, R., Zhang, F., & Nureki, O. (2014). Crystal Structure of Cas9 in Complex with Guide RNA and Target DNA. Cell, 156(5), 935–949. https://doi.org/10.1016/j.cell.2014.02.001 PMID: 24529477
Nishimasu, H., Shi, X., Ishiguro, S., Gao, L., Hirano, S., Okazaki, S., Noda, T., Abudayyeh, O. O., Gootenberg, J. S., Mori, H., Oura, S., Holmes, B., Tanaka, M., Seki, M., Hirano, H., Aburatani, H., Ishitani, R., Ikawa, M., Yachie, N., . . . Nureki, O. (2018). Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science, 361(6408), 1259–1262. https://doi.org/10.1126/science.aas9129 PMID: 30166441
Pawluk, A., Amrani, N., Zhang, Y., Garcia, B., Hidalgo-Reyes, Y., Lee, J., Edraki, A., Shah, M., Sontheimer, E. J., Maxwell, K. L., & Davidson, A. R. (2016). Naturally occurring Off-Switches for CRISPR-Cas9. Cell, 167(7), 1829-1838.e9. https://doi.org/10.1016/j.cell.2016.11.017 PMID: 27984730
Ran, F. A., Hsu, P. D., Wright, J., Agarwala, V., Scott, D. A., & Zhang, F. (2013). Genome engineering using the CRISPR-Cas9 system. Nature Protocols, 8(11), 2281–2308. https://doi.org/10.1038/nprot.2013.143 PMID: 24157548
Ran, F. A., Cong, L., Yan, W. X., Scott, D. A., Gootenberg, J. S., Kriz, A. J., Zetsche, B., Shalem, O., Wu, X., Makarova, K. S., Koonin, E. V., Sharp, P. A., & Zhang, F. (2015). In vivo genome editing using Staphylococcus aureus Cas9. Nature, 520(7546), 186–191. https://doi.org/10.1038/nature14299 PMID: 25830891
Rauch, B. J., Silvis, M. R., Hultquist, J. F., Waters, C. S., McGregor, M. J., Krogan, N. J., & Bondy-Denomy, J. (2017). Inhibition of CRISPR-Cas9 with Bacteriophage Proteins. Cell, 168(1–2), 150-158.e10. https://doi.org/10.1016/j.cell.2016.12.009 PMID: 28041849
Sakuma, T., Nishikawa, A., Kume, S., Chayama, K., & Yamamoto, T. (2014). Multiplex genome engineering in human cells using all-in-one CRISPR/Cas9 vector system. Scientific Reports, 4(1). https://doi.org/10.1038/srep05400 PMID: 24954249
Shechner, D. M., Hacisuleyman, E., Younger, S. T., & Rinn, J. L. (2015). Multiplexable, locus-specific targeting of long RNAs with CRISPR-Display. Nature Methods, 12(7), 664–670. https://doi.org/10.1038/nmeth.3433 PMID: 26030444
Slaymaker, I. M., Gao, L., Zetsche, B., Scott, D. A., Yan, W. X., & Zhang, F. (2016). Rationally engineered Cas9 nucleases with improved specificity. Science, 351(6268), 84–88. https://doi.org/10.1126/science.aad5227 PMID: 26628643
Trevino, A. E., & Zhang, F. (2014). Genome editing using CAS9 Nickases. Methods in Enzymology, 546, 161–174. https://doi.org/10.1016/B978-0-12-801185-0.00008-8 PMID: 25398340
Walton, R. T., Christie, K. A., Whittaker, M. N., & Kleinstiver, B. P. (2020). Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science, 368(6488), 290–296. https://doi.org/10.1126/science.aba8853 PMID: 32217751
Wang, H., Yang, H., Shivalila, C. S., Dawlaty, M. M., Cheng, A. W., Zhang, F., & Jaenisch, R. (2013). One-Step generation of mice carrying mutations in multiple genes by CRISPR/CAS-Mediated Genome Engineering. Cell, 153(4), 910–918. https://doi.org/10.1016/j.cell.2013.04.025 PMID: 23643243
Zhang, Y., & Marchisio, M. A. (2020). Type II anti-CRISPR proteins as a new tool for synthetic biology. RNA Biology, 18(8), 1085–1098. https://doi.org/10.1080/15476286.2020.1827803 PMID: 32991234
Zhao, L., Koseki, S. R. T., Silverstein, R. A., Amrani, N., Peng, C., Kramme, C., Savic, N., Pacesa, M., Rodríguez, T. C., Stan, T., Tysinger, E., Hong, L., Yudistyra, V., Ponnapati, M. R., Jacobson, J. M., Church, G. M., Jakimo, N., Truant, R., Jinek, M., . . . Chatterjee, P. (2023). PAM-flexible genome editing with an engineered chimeric Cas9. Nature Communications, 14(1), 6175. https://doi.org/10.1038/s41467-023-41829-y PMID: 37794046
Small precision edits
Anzalone, A. V., Randolph, P. B., Davis, J. R., Sousa, A. A., Koblan, L. W., Levy, J. M., Chen, P. J., Wilson, C., Newby, G. A., Raguram, A., & Liu, D. R. (2019). Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 576(7785), 149–157. https://doi.org/10.1038/s41586-019-1711-4 PMID: 31634902
Anzalone, A. V., Koblan, L. W., & Liu, D. R. (2020). Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nature Biotechnology. 38(7), 824–844. https://doi.org/10.1038/s41587-020-0561-9 PMID: 32572269
Charpentier, M., Khedher, A. H. Y., Menoret, S., Brion, A., Lamribet, K., Dardillac, E., Boix, C., Perrouault, L., Tesson, L., Geny, S., De Cian, A., Itier, J. M., Anegon, I., Lopez, B., Giovannangeli, C., & Concordet, J. P. (2018). CtIP fusion to Cas9 enhances transgene integration by homology-dependent repair. Nature Communications. 9(1), 1133. https://doi.org/10.1038/s41467-018-03475-7 PMID: 29556040
Chen, P. J., Hussmann, J. A., Yan, J., Knipping, F., Ravisankar, P., Chen, P., Chen, C., Nelson, J. W., Newby, G. A., Sahin, M., Osborn, M. J., Weissman, J. S., Adamson, B., & Liu, D. R. (2021). Enhanced prime editing systems by manipulating cellular determinants of editing outcomes. Cell. 184(22), 5635-5652.e29. https://doi.org/10.1016/j.cell.2021.09.018 PMID: 34653350
Chu, V. T., Weber, T., Wefers, B., Wurst, W., Sander, S., Rajewsky, K., & Kühn, R. (2015). Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nature Biotechnology. 33(5), 543–548. https://doi.org/10.1038/nbt.3198 PMID: 25803306
Doman, J. L., Pandey, S., Neugebauer, M. E., An, M., Davis, J. R., Randolph, P. B., McElroy, A., Gao, X. D., Raguram, A., Richter, M. F., Everette, K. A., Banskota, S., Tian, K., Tao, Y. A., Tolar, J., Osborn, M. J., & Liu, D. R. (2023). Phage-assisted evolution and protein engineering yield compact, efficient prime editors. Cell. 186(18), 3983-4002.e26. https://doi.org/10.1016/j.cell.2023.07.039 PMID: 37657419
Eid, A., Alshareef, S., & Mahfouz, M. M. (2018). CRISPR base editors: genome editing without double-stranded breaks. Biochemical Journal. 475(11), 1955–1964. https://doi.org/10.1042/BCJ20170793 PMID: 29891532
Gaudelli, N. M., Komor, A. C., Rees, H. A., Packer, M. S., Badran, A. H., Bryson, D. I., & Liu, D. R. (2017). Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature. 551(7681), 464–471. https://doi.org/10.1038/nature24644 PMID: 29160308
Hess, G. T., Frésard, L., Han, K., Lee, C. H., Li, A., Cimprich, K. A., Montgomery, S. B., & Bassik, M. C. (2016). Directed evolution using dCas9-targeted somatic hypermutation in mammalian cells. Nature Methods. 13(12), 1036–1042. https://doi.org/10.1038/nmeth.4038 PMID: 27798611
Kim, Y. B., Komor, A. C., Levy, J. M., Packer, M. S., Zhao, K. T., & Liu, D. R. (2017). Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nature Biotechnology. 35(4), 371–376. https://doi.org/10.1038/nbt.3803 PMID: 28191901
Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A., & Liu, D. R. (2016). Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 533(7603), 420–424. https://doi.org/10.1038/nature17946 PMID: 27096365
Komor, A. C., Zhao, K. T., Packer, M. S., Gaudelli, N. M., Waterbury, A. L., Koblan, L. W., Kim, Y. B., Badran, A. H., & Liu, D. R. (2017). Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. Science Advances. 3(8), eaao4774. https://doi.org/10.1126/sciadv.aao4774 PMID: 28875174
Leal, A., Herreno-Pachón, A., Benincore-Flórez, E., Karunathilaka, A., & Tomatsu, S. (2024). Current strategies for increasing Knock-In efficiency in CRISPR/CAS9-Based approaches. International Journal of Molecular Sciencess. 25(5), 2456. https://doi.org/10.3390/ijms25052456 PMID: 38473704
Lin, S., Staahl, B. T., Alla, R. K., & Doudna, J. A. (2014). Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife. 3e04766. https://doi.org/10.7554/eLife.04766 PMID: 25497837
Ma, Y., Zhang, J., Yin, W., Zhang, Z., Song, Y., & Chang, X. (2016). Targeted AID-mediated mutagenesis (TAM) enables efficient genomic diversification in mammalian cells. Nature Methods. 13(12), 1029–1035. https://doi.org/10.1038/nmeth.4027 PMID: 27723754
Rees, H. A., Komor, A. C., Yeh, W., Caetano-Lopes, J., Warman, M., Edge, A. S. B., & Liu, D. R. (2017). Improving the DNA specificity and applicability of base editing through protein engineering and protein delivery. Nature Communications. 8, 15790. https://doi.org/10.1038/ncomms15790 PMID: 28585549
Rees, H. A., & Liu, D. R. (2018). Base editing: precision chemistry on the genome and transcriptome of living cells. Nature Reviews Genetics. 19(12), 770–788. https://doi.org/10.1038/s41576-018-0059-1 PMID: 30323312
Yan, J., Oyler-Castrillo, P., Ravisankar, P., Ward, C. C., Levesque, S., Jing, Y., Simpson, D., Zhao, A., Li, H., Yan, W., Goudy, L., Schmidt, R., Solley, S. C., Gilbert, L. A., Chan, M. M., Bauer, D. E., Marson, A., Parsons, L. R., & Adamson, B. (2024). Improving prime editing with an endogenous small RNA-binding protein. Nature. 628(8008), 639–647. https://doi.org/10.1038/s41586-024-07259-6 PMID: 38570691
Large scale edits
Chen, S. P., & Wang, H. H. (2019). An engineered CaS-Transposon system for programmable and Site-Directed DNA transpositions. The CRISPR Journal. 2(6), 376–394. https://doi.org/10.1089/crispr.2019.0030 PMID: 31742433
Durrant, M. G., Fanton, A., Tycko, J., Hinks, M., Chandrasekaran, S. S., Perry, N. T., Schaepe, J., Du, P. P., Lotfy, P., Bassik, M. C., Bintu, L., Bhatt, A. S., & Hsu, P. D. (2022). Systematic discovery of recombinases for efficient integration of large DNA sequences into the human genome. Nature Biotechnology. 41(4), 488–499. https://doi.org/10.1038/s41587-022-01494-w PMID: 36217031
Gelsinger, D. R., Vo, P. L. H., Klompe, S. E., Ronda, C., Wang, H. H., & Sternberg, S. H. (2024). Bacterial genome engineering using CRISPR-associated transposases. Nature Protocols. 19(3), 752–790. https://doi.org/10.1038/s41596-023-00927-3 PMID: 38216671
Klompe, S. E., Vo, P. L. H., Halpin-Healy, T. S., & Sternberg, S. H. (2019). Transposon-encoded CRISPR–Cas systems direct RNA-guided DNA integration. Nature. 571(7764), 219–225. https://doi.org/10.1038/s41586-019-1323-z PMID: 31189177
Strecker, J., Ladha, A., Gardner, Z., Schmid-Burgk, J. L., Makarova, K. S., Koonin, E. V., & Zhang, F. (2019). RNA-guided DNA insertion with CRISPR-associated transposases. Science. 365(6448), 48–53. https://doi.org/10.1126/science.aax9181 PMID: 31171706
Tou, C. J., Orr, B., & Kleinstiver, B. P. (2023). Precise cut-and-paste DNA insertion using engineered type V-K CRISPR-associated transposases. Nature Biotechnology. 41(7), 968–979. https://doi.org/10.1038/s41587-022-01574-x PMID: 36593413
Vo, P. L. H., Ronda, C., Klompe, S. E., Chen, E. E., Acree, C., Wang, H. H., & Sternberg, S. H. (2020). CRISPR RNA-guided integrases for high-efficiency, multiplexed bacterial genome engineering. Nature Biotechnology. 39(4), 480–489. https://doi.org/10.1038/s41587-020-00745-y PMID: 33230293
Yang, S., Zhang, Y., Xu, J., Zhang, J., Zhang, J., Yang, J., Jiang, Y., & Yang, S. (2021). Orthogonal CRISPR-associated transposases for parallel and multiplexed chromosomal integration. Nucleic Acids Research. 49(17), 10192–10202. https://doi.org/10.1093/nar/gkab752 PMID: 34478496
Yarnall, M. T. N., Ioannidi, E. I., Schmitt-Ulms, C., Krajeski, R. N., Lim, J., Villiger, L., Zhou, W., Jiang, K., Garushyants, S. K., Roberts, N., Zhang, L., Vakulskas, C. A., Walker, J. A., Kadina, A. P., Zepeda, A. E., Holden, K., Ma, H., Xie, J., Gao, G., . . . Gootenberg, J. S. (2022). Drag-and-drop genome insertion of large sequences without double-strand DNA cleavage using CRISPR-directed integrases. Nature Biotechnology. 41(4), 500–512. https://doi.org/10.1038/s41587-022-01527-4 PMID: 36424489
Genome-wide screening
McDade, J. R., Waxmonsky, N. C., Swanson, L. E., & Fan, M. (2016). Practical considerations for using pooled lentiviral CRISPR libraries. Current Protocols in Molecular Biology. 115. https://doi.org/10.1002/cpmb.8 PMID: 27366891
Rousset, F., Cui, L., Siouve, E., Becavin, C., Depardieu, F., & Bikard, D. (2018). Genome-wide CRISPR-dCas9 screens in E. coli identify essential genes and phage host factors. PLOS Genetics. 14(11), e1007749. https://doi.org/10.1371/journal.pgen.1007749 PMID: 30403660
Sanson, K. R., Hanna, R. E., Hegde, M., Donovan, K. F., Strand, C., Sullender, M. E., Vaimberg, E. W., Goodale, A., Root, D. E., Piccioni, F., & Doench, J. G. (2018). Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nature Communications. 9(11), e1007749. https://doi.org/10.1038/s41467-018-07901-8 PMID: 30575746
Shalem, O., Sanjana, N. E., Hartenian, E., Shi, X., Scott, D. A., Mikkelsen, T. S., Heckl, D., Ebert, B. L., Root, D. E., Doench, J. G., & Zhang, F. (2014). Genome-Scale CRISPR-CAS9 knockout screening in human cells. Science. 343(6166), 84–87. https://doi.org/10.1126/science.1247005 PMID: 24336571
Shalem, O., Sanjana, N. E., & Zhang, F. (2015). High-throughput functional genomics using CRISPR–Cas9. Nature Reviews Genetics. 16(5), 299–311. https://doi.org/10.1038/nrg3899 PMID: 25854182
Shi, J., Wang, E., Milazzo, J. P., Wang, Z., Kinney, J. B., & Vakoc, C. R. (2015). Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nature Biotechnology. 33(6), 661–667. https://doi.org/10.1038/nbt.3235 PMID: 25961408
Wang, T., Wei, J. J., Sabatini, D. M., & Lander, E. S. (2014). Genetic screens in human cells using the CRISPR-CAS9 system. Science. 343(6166), 80–84. https://doi.org/10.1126/science.1246981 PMID: 24336569
Zhong, C., Yin, Q., Xie, Z., Bai, M., Dong, R., Tang, W., Xing, Y., Zhang, H., Yang, S., Chen, L., Bartolomei, M. S., Ferguson-Smith, A., Li, D., Yang, L., Wu, Y., & Li, J. (2015). CRISPR-Cas9-Mediated Genetic Screening in Mice with Haploid Embryonic Stem Cells Carrying a Guide RNA Library. Cell Stem Cell. 17(2), 221–232. https://doi.org/10.1016/j.stem.2015.06.005 PMID: 26165924
Cas9 fusion tools
Chavez, A., Scheiman, J., Vora, S., Pruitt, B. W., Tuttle, M., Iyer, E. P. R., Lin, S., Kiani, S., Guzman, C. D., Wiegand, D. J., Ter-Ovanesyan, D., Braff, J. L., Davidsohn, N., Housden, B. E., Perrimon, N., Weiss, R., Aach, J., Collins, J. J., & Church, G. M. (2015). Highly efficient Cas9-mediated transcriptional programming. Nature Methods. 12(4), 326–328. https://doi.org/10.1038/nmeth.3312 PMID: 25730490
Chen, B., Gilbert, L. A., Cimini, B. A., Schnitzbauer, J., Zhang, W., Li, G., Park, J., Blackburn, E. H., Weissman, J. S., Qi, L. S., & Huang, B. (2013). Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/CAS system. Cell. 155(7), 1479–1491. https://doi.org/10.1016/j.cell.2013.12.001 PMID: 24360272
Dalvai, M., Loehr, J., Jacquet, K., Huard, C. C., Roques, C., Herst, P., Côté, J., & Doyon, Y. (2015). A scalable Genome-Editing-Based approach for mapping multiprotein complexes in human cells. Cell Reports. 13(3), 621–633. https://doi.org/10.1016/j.celrep.2015.09.009 PMID: 26456817
Doench, J. G., Hartenian, E., Graham, D. B., Tothova, Z., Hegde, M., Smith, I., Sullender, M., Ebert, B. L., Xavier, R. J., & Root, D. E. (2014). Rational design of highly active sgRNAs for CRISPR-Cas9–mediated gene inactivation. Nature Biotechnology. 32(12), 1262–1267. https://doi.org/10.1038/nbt.3026 PMID: 25184501
Dong, C., Fontana, J., Patel, A., Carothers, J. M., & Zalatan, J. G. (2018). Synthetic CRISPR-Cas gene activators for transcriptional reprogramming in bacteria. Nature Communications. 9(1), 2489. https://doi.org/10.1038/s41467-018-04901-6 PMID: 29950558
Fujita, T., & Fujii, H. (2013). Efficient isolation of specific genomic regions and identification of associated proteins by engineered DNA-binding molecule-mediated chromatin immunoprecipitation (enChIP) using CRISPR. Biochemical and Biophysical Research Communications. 439(1), 132–136. https://doi.org/10.1016/j.bbrc.2013.08.013 PMID: 23942116
Gilbert, L. A., Larson, M. H., Morsut, L., Liu, Z., Brar, G. A., Torres, S. E., Stern-Ginossar, N., Brandman, O., Whitehead, E. H., Doudna, J. A., Lim, W. A., Weissman, J. S., & Qi, L. S. (2013). CRISPR-Mediated modular RNA-Guided regulation of transcription in eukaryotes. Cell. 154(2), 442–451. https://doi.org/10.1016/j.cell.2013.06.044 PMID: 23849981
Gilbert, L. A., Horlbeck, M. A., Adamson, B., Villalta, J. E., Chen, Y., Whitehead, E. H., Guimaraes, C., Panning, B., Ploegh, H. L., Bassik, M. C., Qi, L. S., Kampmann, M., & Weissman, J. S. (2014). Genome-Scale CRISPR-Mediated control of gene repression and activation. Cell. 159(3), 647–661. https://doi.org/10.1016/j.cell.2014.09.029 PMID: 25307932
Hilton, I. B., D’Ippolito, A. M., Vockley, C. M., Thakore, P. I., Crawford, G. E., Reddy, T. E., & Gersbach, C. A. (2015). Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nature Biotechnology. 33(5), 510–517. https://doi.org/10.1038/nbt.3199 PMID: 25849900
Konermann, S., Brigham, M. D., Trevino, A. E., Joung, J., Abudayyeh, O. O., Barcena, C., Hsu, P. D., Habib, N., Gootenberg, J. S., Nishimasu, H., Nureki, O., & Zhang, F. (2015). Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 517(7536), 583–588. https://doi.org/10.1038/nature14136 PMID: 25494202
Liu, X., Zhang, Y., Chen, Y., Li, M., Zhou, F., Li, K., Cao, H., Ni, M., Liu, Y., Gu, Z., Dickerson, K. E., Xie, S., Hon, G. C., Xuan, Z., Zhang, M. Q., Shao, Z., & Xu, J. (2017). In situ capture of chromatin interactions by biotinylated DCAS9. Cell. 170(5), 1028-1043.e19. https://doi.org/10.1016/j.cell.2017.08.003 PMID: 28841410
Ma, H., Naseri, A., Reyes-Gutierrez, P., Wolfe, S. A., Zhang, S., & Pederson, T. (2015). Multicolor CRISPR labeling of chromosomal loci in human cells. PNAS. 112(10), 3002–3007. https://doi.org/10.1073/pnas.1420024112 PMID: 25713381
Ma, H., Tu, L., Naseri, A., Huisman, M., Zhang, S., Grunwald, D., & Pederson, T. (2016). Multiplexed labeling of genomic loci with dCas9 and engineered sgRNAs using CRISPRainbow. Nature Biotechnology. 34(5), 528–530. https://doi.org/10.1038/nbt.3526 PMID: 27088723
Ma, H., Tu, L., Naseri, A., Chung, Y., Grunwald, D., Zhang, S., & Pederson, T. (2018). CRISPR-Sirius: RNA scaffolds for signal amplification in genome imaging. Nature Methodss. 15(11), 928–931. https://doi.org/10.1038/s41592-018-0174-0 PMID: 30377374
Mali, P., Aach, J., Stranges, P. B., Esvelt, K. M., Moosburner, M., Kosuri, S., Yang, L., & Church, G. M. (2013). CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nature Biotechnology. 31(9), 833–838. https://doi.org/10.1038/nbt.2675 PMID: 23907171
Nuñez, J. K., Chen, J., Pommier, G. C., Cogan, J. Z., Replogle, J. M., Adriaens, C., Ramadoss, G. N., Shi, Q., Hung, K. L., Samelson, A. J., Pogson, A. N., Kim, J. Y., Chung, A., Leonetti, M. D., Chang, H. Y., Kampmann, M., Bernstein, B. E., Hovestadt, V., Gilbert, L. A., & Weissman, J. S. (2021). Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing. Cell. 184(9), 2503-2519.e17. https://doi.org/10.1016/j.cell.2021.03.025 PMID: 33838111
Peters, J. M., Koo, B., Patino, R., Heussler, G. E., Hearne, C. C., Qu, J., Inclan, Y. F., Hawkins, J. S., Lu, C. H. S., Silvis, M. R., Harden, M. M., Osadnik, H., Peters, J. E., Engel, J. N., Dutton, R. J., Grossman, A. D., Gross, C. A., & Rosenberg, O. S. (2019). Enabling genetic analysis of diverse bacteria with Mobile-CRISPRi. Nature Microbiology. 4(2), 244–250. https://doi.org/10.1038/s41564-018-0327-z PMID: 30617347
Qi, L. S., Larson, M. H., Gilbert, L. A., Doudna, J. A., Weissman, J. S., Arkin, A. P., & Lim, W. A. (2013). Repurposing CRISPR as an RNA-Guided platform for Sequence-Specific control of gene expression. Cell. 152(5), 1173–1183. https://doi.org/10.1016/j.cell.2013.02.022 PMID: 23452860
Savic, D., Partridge, E. C., Newberry, K. M., Smith, S. B., Meadows, S. K., Roberts, B. S., Mackiewicz, M., Mendenhall, E. M., & Myers, R. M. (2015). CETCh-seq: CRISPR epitope tagging ChIP-seq of DNA-binding proteins. Genome Research. 25(10), 1581–1589. https://doi.org/10.1101/gr.193540.115 PMID: 26355004
Tanenbaum, M. E., Gilbert, L. A., Qi, L. S., Weissman, J. S., & Vale, R. D. (2014). A Protein-Tagging system for signal amplification in gene expression and fluorescence imaging. Cell. 159(3), 635–646. https://doi.org/10.1016/j.cell.2014.09.039 PMID: 25307933
Zalatan, J. G., Lee, M. E., Almeida, R., Gilbert, L. A., Whitehead, E. H., La Russa, M., Tsai, J. C., Weissman, J. S., Dueber, J. E., Qi, L. S., & Lim, W. A. (2015). Engineering Complex Synthetic Transcriptional Programs with CRISPR RNA Scaffolds. Cell. 160(1–2), 339–350. https://doi.org/10.1016/j.cell.2014.11.052 PMID: 25533786
Cas9 alternatives
Abudayyeh, O. O., Gootenberg, J. S., Essletzbichler, P., Han, S., Joung, J., Belanto, J. J., Verdine, V., Cox, D. B. T., Kellner, M. J., Regev, A., Lander, E. S., Voytas, D. F., Ting, A. Y., & Zhang, F. (2017). RNA targeting with CRISPR–Cas13. Nature, 550(7675), 280–284. https://doi.org/10.1038/nature24049 PMID: 28976959
Cox, D. B. T., Gootenberg, J. S., Abudayyeh, O. O., Franklin, B., Kellner, M. J., Joung, J., & Zhang, F. (2017). RNA editing with CRISPR-Cas13. Science, 358(6366), 1019–1027. https://doi.org/10.1126/science.aaq0180 PMID: 29070703
Csörgő, B., León, L. M., Chau-Ly, I. J., Vasquez-Rifo, A., Berry, J. D., Mahendra, C., Crawford, E. D., Lewis, J. D., & Bondy-Denomy, J. (2020). A compact Cascade–Cas3 system for targeted genome engineering. Nature Methods, 17(12), 1183–1190. https://doi.org/10.1038/s41592-020-00980-w PMID: 33077967
DeWeirdt, P. C., Sanson, K. R., Sangree, A. K., Hegde, M., Hanna, R. E., Feeley, M. N., Griffith, A. L., Teng, T., Borys, S. M., Strand, C., Joung, J. K., Kleinstiver, B. P., Pan, X., Huang, A., & Doench, J. G. (2020). Optimization of AsCas12a for combinatorial genetic screens in human cells. Nature Biotechnology, 31(1), 94–104. https://doi.org/10.1038/s41587-020-0600-6 PMID: 32661438
Gao, L., Cox, D. B. T., Yan, W. X., Manteiga, J. C., Schneider, M. W., Yamano, T., Nishimasu, H., Nureki, O., Crosetto, N., & Zhang, F. (2017). Engineered Cpf1 variants with altered PAM specificities. Nature Biotechnology, 35(8), 789–792. https://doi.org/10.1038/nbt.3900 PMID: 28581492
Gootenberg, J. S., Abudayyeh, O. O., Lee, J. W., Essletzbichler, P., Dy, A. J., Joung, J., Verdine, V., Donghia, N., Daringer, N. M., Freije, C. A., Myhrvold, C., Bhattacharyya, R. P., Livny, J., Regev, A., Koonin, E. V., Hung, D. T., Sabeti, P. C., Collins, J. J., & Zhang, F. (2017). Nucleic acid detection with CRISPR-Cas13a/C2c2. Science, 356(6336), 438–442. https://doi.org/10.1126/science.aam9321 PMID: 28408723
Kleinstiver, B. P., Sousa, A. A., Walton, R. T., Tak, Y. E., Hsu, J. Y., Clement, K., Welch, M. M., Horng, J. E., Malagon-Lopez, J., Scarfò, I., Maus, M. V., Pinello, L., Aryee, M. J., & Joung, J. K. (2019). Engineered CRISPR–Cas12a variants with increased activities and improved targeting ranges for gene, epigenetic and base editing. Nature Biotechnology, 37(3), 276–282. https://doi.org/10.1038/s41587-018-0011-0 PMID: 30742127
Loeff, L., Brouns, S. J., & Joo, C. (2018). Repetitive DNA reeling by the Cascade-Cas3 complex in nucleotide unwinding steps. Molecular Cell, 70(3), 385-394.e3. https://doi.org/10.1016/j.molcel.2018.03.031 PMID: 29706536
Morisaka, H., Yoshimi, K., Okuzaki, Y., Gee, P., Kunihiro, Y., Sonpho, E., Xu, H., Sasakawa, N., Naito, Y., Nakada, S., Yamamoto, T., Sano, S., Hotta, A., Takeda, J., & Mashimo, T. (2019). CRISPR-Cas3 induces broad and unidirectional genome editing in human cells. Nature Communications, 10(1), 5302. https://doi.org/10.1038/s41467-019-13226-x PMID: 31811138
O’Brien, R. E., Bravo, J. P., Ramos, D., Hibshman, G. N., Wright, J. T., & Taylor, D. W. (2023). Structural snapshots of R-loop formation by a type I-C CRISPR Cascade. Molecular Cell, 83(5), 746-758.e5. https://doi.org/10.1016/j.molcel.2023.01.024 PMID: 36805026
Terns, M. P. (2018). CRISPR-Based Technologies: Impact of RNA-Targeting Systems. Molecular Cell, 72(3), 404–412. https://doi.org/10.1016/j.molcel.2018.09.018 PMID: 30388409
Westra, E. R., Van Erp, P. B., Künne, T., Wong, S. P., Staals, R. H., Seegers, C. L., Bollen, S., Jore, M. M., Semenova, E., Severinov, K., De Vos, W. M., Dame, R. T., De Vries, R., Brouns, S. J., & Van Der Oost, J. (2012). CRISPR immunity relies on the consecutive binding and degradation of negatively supercoiled invader DNA by cascade and CAS3. Molecular Cell, 46(5), 595–605. https://doi.org/10.1016/j.molcel.2012.03.018 PMID: 22521689
Yoshimi, K., & Mashimo, T. (2022). Genome editing technology and applications with the type I CRISPR system. Gene and Genome Editing, 3–4, 100013. https://doi.org/10.1016/j.ggedit.2022.100013 PMID: 10.1016/j.ggedit.2022.100013
Zetsche, B., Gootenberg, J. S., Abudayyeh, O. O., Slaymaker, I. M., Makarova, K. S., Essletzbichler, P., Volz, S. E., Joung, J., Van Der Oost, J., Regev, A., Koonin, E. V., & Zhang, F. (2015). CPF1 is a single RNA-Guided endonuclease of a Class 2 CRISPR-CAS system. Cell, 163(3), 759–771. https://doi.org/10.1016/j.cell.2015.09.038 PMID: 26422227
Zetsche, B., Heidenreich, M., Mohanraju, P., Fedorova, I., Kneppers, J., DeGennaro, E. M., Winblad, N., Choudhury, S. R., Abudayyeh, O. O., Gootenberg, J. S., Wu, W. Y., Scott, D. A., Severinov, K., Van Der Oost, J., & Zhang, F. (2017). Multiplex gene editing by CRISPR–Cpf1 using a single crRNA array. Nature Biotechnology, 35(1), 31–34. https://doi.org/10.1038/nbt.3737 PMID: 27918548